

应同写意邀请,国家癌症中心/中国医学科学院肿瘤医院副院长李宁在“2023年中国药学会药事管理学术年会暨首届苏州未来新药大会”上做了关于《中国医药创新的临床需求和临床能力》报告,本文系根据报告内容整理。

李宁
国家癌症中心/中国医学科学院肿瘤医院副院长
1
医药创新:成果显著
从肿瘤来说,国内的临床需求非常大,对整个创新药和市场都有很大的需求。一项数据显示,中国新发肿瘤病例数全球第一,占全球23.9%;但5年生存率较美国相对低(41% vs 65%)。有限的治疗药物是造成患者预后不佳的主要因素。
为什么会有这么大的差别呢?不是中国医生不努力,很大程度上来说,过去中国治疗和国际领先的疗法相比,曾经差了一到两个时代。在2016年之前,中国新药从提交申请到审批平均需要7年,从审批到临床平均需要25个月,从上市到进入医保又平均要7到8年。
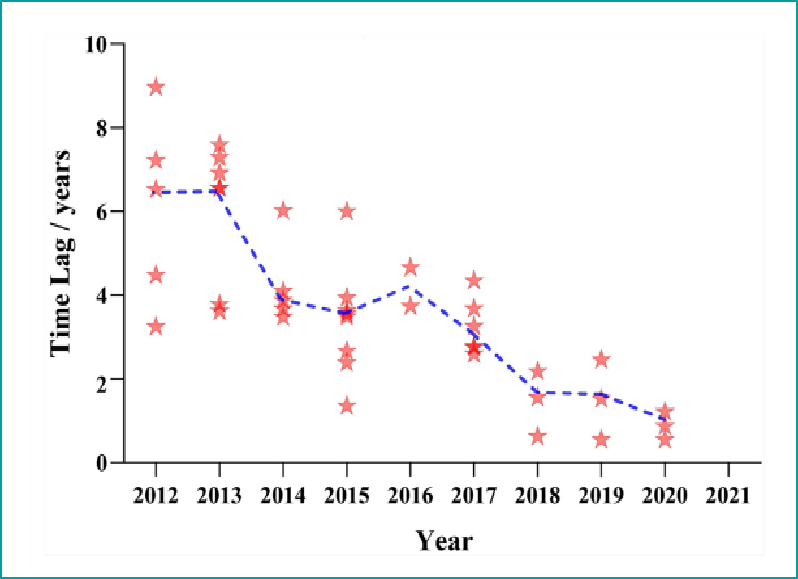
过去十年,中国药物研发表现出强大的活力,取得了巨大的进展,药物可获得性大幅提升、上市时间差也显著缩短。如图所示,原先国际的“大咖”药物可能需要七八年的时间才能进入中国,到2020年时,所需的时间已经非常短了,这些都得益于国内体制和机制的改革。
从获益上来讲,国内疗法也取得了显著进步。大家都觉得布局PD-1的企业太多了,这个靶点出来的时间也很长了。但我想说,中国的创新药企、临床研究者大多的创新意识正是在“PD-1时代”开始的。这之后,中国也确实有了属于自己的创新产品。
另一个突破是药物的进步。在化疗时代,以肺癌为例,五年存活率始终徘徊在12.7%。如今,得益于靶向治疗和免疫治疗的出现,存活率有了巨大的提高,2023年我国肺癌5年生存率是19.7%。对于80多万的肺癌年发病率来讲,7%的改善意味着每年超过5万人的获益。这是医药创新研发在临床上使患者获益的实例。
中国的临床研发时常被诟病都是“me-too药”,但是“me-too药”也具有价值,也能推进中国的研究、推动行业的改革、推进历史的发展,使患者有药可用、用得起药,国内的价格比国际上要低很多。药价低虽说可能不利于研发,但是对患者来说是获益的,这是我们对社会的付出。
即便如此,我们依然没有达到足够的创新,还有很多东西可以做。比如CGT研究,在这方面中国实际上已经走在了比较靠前的阶段,我们拥有独创的“双轨制”模式。使得细胞治疗可以通过医疗技术的方式更加快捷的进入临床实践探究,但在最后成药的阶段仍需要通过药品监管模式完成审批注册。我们仍然还有很多努力需要尝试。
对于创新药物的接受和理解,只有医生、CMO理解还远远不够,我们需要全社会的认同和鼓励。在美国,通过对第一个接受CAR-T治疗的患者Emily进行跟踪报道,公众对CGT的认可程度很高,但在中国,早期细胞治疗个例负面消息的传播使得CGT行业需要更多的准备才能改变公众的认知现状。
科学家们做了很多努力将这个技术推到临床,却没有患者相信,这种情况非常难以继续开展研究,我们有必要拿出一些优秀的例子向社会进行反馈,让大家意识到临床研究、创新治疗的意义。
2
创新能力:时代需求
这个时代对中国的创新提出了新要求,me too、me better、fast follow产品我们都已经达到了很高的水平,接下来要自己往前走了。可喜的是,国内的资源也确实给自主创新提供了很多条件。
首先是政策上的支持。2017年至今,CDE颁布药品技术指导原则311部。相当于每周末都要发布指导原则,CDE努力地想让大家更了解政策。其次,加入ICH的意义也很非凡。要出海就要达到这种标准,政策上有支持,基础上也要有沉淀。
2023自然指数年度榜单发布,中国对自然科学贡献首超美国居首位,说明我们的基础沉淀也在日益提高。有很多厉害的科学家们愿意去创业,科学家们做的东西是奔着转化去的,在这方面政策上也有很多支持。国家卫健委重大新药创制计划,十一五到十三五,中央财政经费投入超过200亿,支持项目3000余项,产出上市一类新药85个,实现国产创新药在欧美日等发达国家上市,主营收入超百亿的企业从专项实施前的两家发展到超过20家。
医院部分也有很多配合。北京建设了很多研究性病房,苏州、上海在做研究性医院,医院里面的研究者们也愿意出来跟大家沟通了,这是各方积极配合的一个结果。
临床研究的质量是永恒的目标,没有上限的要求。FDA现场核查的标准有3个:NAI(不需要采取行政措施)、VAI(企业或机构自愿采取行政措施)、OAI(FDA需要采取官方行政措施)。
2016-2023年,FDA共核查中国机构18次,零次OAI。检查结果排在前列,不劣于欧美等相应发达国家。从政策支持,到行业内从源头到实施的充分转化沟通,再到临床研究的质量保障,种种资源已为创新药研究铺平了道路。

3
创新转化:问题何在
每年,全球CNS及子刊发表肿瘤相关临床研究数量超过5000篇,全球发布肿瘤三期确证性临床研究数量超过300个,FDA批准扩展适应症药物超过50个,但FDA批准的新药物不到20个,差不多是1%的几率。
创新是一个风险非常大的领域,但很长时间以来,国内的我们只有成功的经验,没有失败的经验,这并不是因为我们技术好,反而是因为还没有做到位,技术还没有突破。中国临床试验也是一样,只有成功的经验,其实没有失败经验的研究很难取得进一步的成果。但我们的心理建设好像没那么好,一旦研究失败,严重的话整个团队都会解散,但在国外,失败是一件很正常的事。我们似乎不具备承担失败风险的能力。
临床研究需要科学理念,适应症、评价指标等选择都是严谨的科学问题。关于患者优选的问题,FDA的ODAC会议传递了很多非常有价值的技巧。
今年4月的ODAC会议针对奥拉帕利进行了讨论。奥拉帕利作为DNA修复酶PARP抑制剂,在存在同源重组修复 (HRR,包括BRCA1/2、ATM等多种关键蛋白) 缺陷的患者中可以发挥“合成致死效应”。
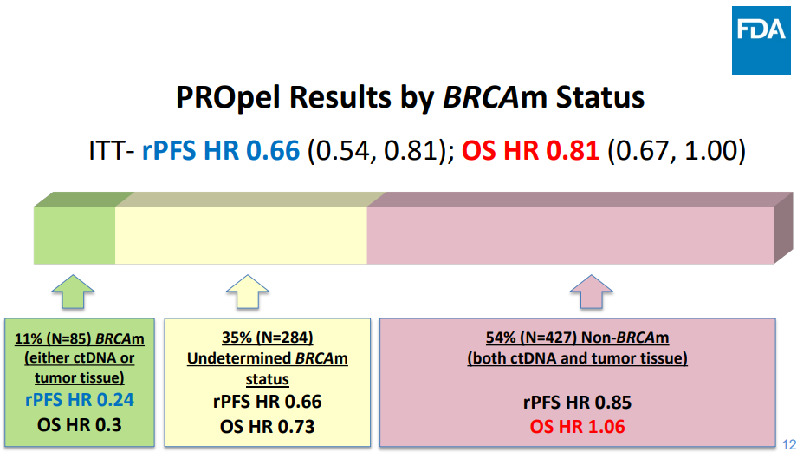
在PROpel临床试验中,阿比特龙+奥拉帕利组和阿比特龙+安慰剂组的随机双盲设计得很完善,结果很好,独立盲审中心 (BICR) 评估结果与研究者评估结果基本一致,研究结果也在《新英格兰杂志》上进行了发表。本该是一个皆大欢喜的结局,但FDA并不这么认为。
FDA将数据整合后重新将患者进行分层,通过研究发现,该疗法只能让一小部分人群获益而非提交申请材料上的“整体获益”,因此认为奥拉帕利联合阿比特龙用于转移性去势抵抗性前列腺癌 (mCRPC) 患者初始治疗的适应症应严格限制在肿瘤伴BRCA突变的患者中。
这个例子表明,只有阳性结果是不够的,还要有科学的理论和理由,不但要拿出临床结果,更要分层结论寻找适合的获益人群。
另一个案例是今年10月的ODAC会议。当时专家组以2票赞同,10票反对的结果,认为sotorasib的III期确证性试验CodeBreaK 200的主要研究终点(BICR的PFS)无法被可靠地解释。与大多数ODAC会议关注研究结果的获益-风险评价不同,此次会议集中于临床试验设计和实施过程中的系统性偏倚问题。

研究达到了主要终点,但关键次要终点OS没有显著差异。投票反对的专家认同没有人期望完美的RCT研究,但希望较大的获益可以承受少量问题带来的不确定性,而CodeBreaK 200研究中一系列问题使得较小的PFS获益变得模糊不清。FDA质疑这个临床结果是否可信。
我非常欣赏这个药物的设计和创新,但是它的研究设计和执行也确实是失败的:没有设计双盲试验,可能是CMO大意了;PI的临床执行上不自主的预设了获益倾向,这使得研究者判断的疾病进展明显倾向实验组,BICR和统计师也都有一些疏忽。创新需要谨慎的心态。
创新是一个非常庞大的体系,除了机制创新,还需要政策创新、评价创新、应用创新、制剂创新、支付体系、社会创新等共同配合。此外,创新也需要专业的分工,从专业到职业发展,以专业的学术精神,职业的使命责任,推动临床研究行业高速发展。
原始创新,意味着独自率先登上了一座基础研究的顶峰。原始创新的临床转化,是遥遥相对的另一座高峰。我相信只要是进入临床III期的药物,没有一个是完全无效的。但寻找合适的方法、合适的剂量,合适的人群、合适的评价体系,把之应用到临床,进行创新转化,则并不容易。各方都需要打好配合,尽可能提高创新的转化率。我们已经遥看到了另一座高峰的雄伟,也希望中国创新药能更多勇攀高峰,收获山巅的风景。
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


