意料之中,医药重大政策周五见,为指导MAH有针对性地开展生物制品上市后药学变更研究,加强生物制品全生命周期管理,确保变更后生物制品的安全性、有效性和质量可控性,2021年6月25日,国家药品监督管理局再放大招,根据《中华人民共和国药品管理法》、《中华人民共和国疫苗管理法》、《药品注册管理办法》、《药品生产监督管理办法》和《药品上市后变更管理办法(试行)》相关规定和要求,CDE官网发布《已上市生物制品药学变更研究技术指导原则(试行)》的通告,自发布之日(2021年6月25日)起施行,一石激起千层浪,对国内生物医药行业具有深远的意义。
这是自16年前,即2005年的《生物制品生产工艺过程变更管理技术指导原则》【国食药监注[2005]493号】发布以来的首次重大修订,生物制品上市后生产工艺变更的监管将成生物制品行业洗牌新“杀器”,生物制品安全,关乎民生,本文梳理了《已上市生物制品药学变更研究技术指导原则(试行)》修订过程中广受社会各界关注的三个热点。
一、已上市生物制品生产工艺变更管理痛点
已上市生物制品生产工艺变更难点是某些
企业非法地要降低成本,监管部门对工艺变更行为缺少可执行的指导原则进行控制,16年前颁布的《生物制品生产工艺过程变更管理技术指导原则》【国食药监注[2005]493号】分类过于笼统且缺少明确的分类原则,也没有提供详细的资料要求,可执行性不够,不能满足现生物制品行业的变更管理需求。
如2018年长生生物疫苗造假案,该企业生产
药品使用的离心机变更未按规定备案及将不同批次的原液进行勾兑配制,再对勾兑合批后的原液重新编造生产批号等违法事实,暴露出部分企业存在对变更研究控制和风险评估管理的重视不够、开展相关研究不充分等问题。
基于上述原因,CDE从全方位完善已上市生物制品生产工艺变更管理为出发点,举一反三,堵塞漏洞,本着鼓励MAH持续改进工艺,提高和保障生物制品质量的目标,组织修订了《已上市生物制品药学变更研究技术指导原则(试行)》。
二、《已上市生物制品药学变更研究技术指导原则(试行)》亮点抢先看
2.1适用范围
2.2变更分类更科学
《已上市生物制品药学变更研究技术指导原则(试行)》中对各变更事项风险评估分级是在基于科学和风险的基础上,按照《中华人民共和国药品管理法》、《中华人民共和国疫苗管理法》、《
药品注册管理办法》、《药品生产监督管理办法》和《药品上市后变更管理办法(试行)》相关规定和要求分为三类,分类的过程中参考了欧美等先进
医药机构发布的相关指导原则,《已上市生物制品药学变更研究技术指导原则(试行)》按药学变更可能对生物制品安全性、有效性和质量可控性的风险和产生影响的程度,实行变更分类。
依据风险和产生影响的程度由高到低分为:重大变更、中等变更、微小变更。
对于重大变更需要通过系列的研究证明,该变更不对产品的安全性、有效性和质量可控性产生不良影响;对于中等变更需要通过相应的研究证明,该变更不影响产品的安全性、有效性,并且不降低产品的质量可控性。
需注意的是中等变更已经取消征求意见稿中的中等变更A、中等变更B了,并且取消了三大类变更的定义,但2020年04月30日发布的《已上市生物制品药学变更研究技术指导原则》(征求意见稿)的定义可作为变更分类评估的参考依据之一。
三、已上市生物制品药学变更申报资料如何与CTD申报相衔接?
已上市生物制品,当发生药学相关变更时,申办者应当充分的评估变更对于受试者安全的影响,在变更前应充分理解产品的性质,评估变更的风险,充分了解
法规要求,生物制品上市后药学变更因生物制品自身特点不同,变更事项不同、变更程度不同,带来的潜在风险也会有所差别。
笔者建议变更相关申报资料可按照《M4:人用药物注册申请通用技术文档(CTD)》中相关模块要求撰写,为了便于申报,对各项常见生物制品药学变更事项标注了其所涉及的通用技术文件(CommonTechnicalDocument,CTD)章节,以与CTD申报相衔接。此次CDE发布《已上市生物制品药学变更研究技术指导原则(试行)》给出了原液和制剂变更分类,为了方便大家查阅。
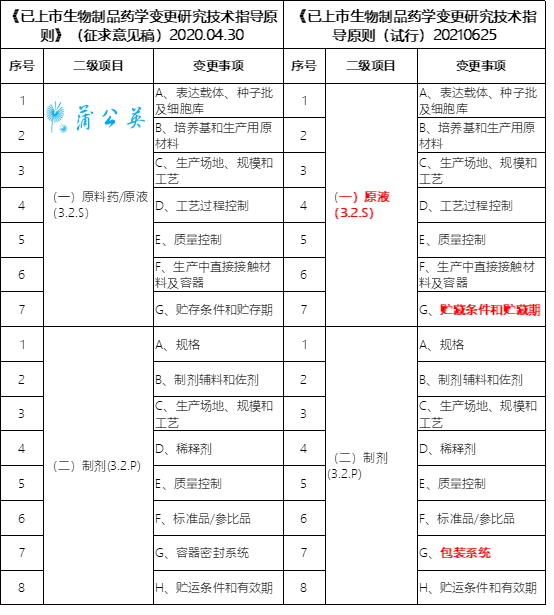
笔者整理对照了2021年06月25日发布实施的《已上市生物制品药学变更研究技术指导原则(试行)》与2020年04月30日发布《已上市生物制品药学变更研究技术指导原则》(征求意见稿)变更对象的差别,主要差别有三点:
(一)原料药/原液(3.2.S)变更为(一)原液(3.2.S)、原液项下G、贮存条件和贮存期变更为G、贮藏条件和贮藏期、制剂项下G、容器密封系统变更为G、包装系统,如下表: