

作者:leixiaolong 丨来源:蒲公英论坛
国内药品生产的基准无疑是药品生产质量管理规范(GMP),该规范的宗旨为防止污染,交叉污染,混淆及差错,污染控制贯穿药品整个生产过程。无菌药品作为药品中质量要求相对较高的一类,其重点就是“无菌”;无菌不合格,不管是因为微生物、微粒、热原外源引入导致还是无菌工艺本身操作控制不当导致,在“无菌药品污染”这个大范围内,亦可理解为其中的一类污染。笔者结合自身无菌药品生产车间经验及对“无菌控制”的理解,浅谈无菌控制的三个难点。
————题记
本文仅代表笔者个人观点,如有错误,还请老师包涵、指正。
无菌生产工艺无菌控制难度>最终灭菌工艺无菌控制难度
众所周知,无菌药品中的无菌是一个概率事件,只是这个概率值需达到官方规则的认可,即无菌保证水平(SAL)=10^-6,含义为10^6灭菌产品中存在活菌的产品不超过1个。
无菌作为无菌药品中重要且具“独特身份标识”的质量属性,通常具有“一票否决权”。一批无菌药品中若检出一支阳性,在没有特别明确的原因下,一般不允许复检,则整个批次会判定存有严重无菌风险,放行面临巨大困难;若调查过程数据及结论不能充分证明无菌不合格的原因,则整批次报废的可能非常大,甚至质量部门会要求重新对无菌保障体系进行验证,合格后才能投入后续生产使用。其实无菌风险每个企业都不愿意承担,毕竟无菌不合格的药品一旦流入市场,会造成严重危害,甚至危及患者生命,对企业本身来讲亦是灭顶之灾。
无菌控制的三大对象为微粒、微生物和热原,其中热原主要指细菌内毒素。防止微生物污染、内毒素污染一直是无菌药品生产企业、监管机构关注的重点。
无菌药品的生产工艺一般分为最终灭菌工艺和无菌生产工艺,后者主要针对热不稳定药品;从过程控制的难易及灭菌彻底性考虑,后者的无菌控制难度远大于前者。
将联系无菌生产工艺生产实际,将无菌风险控制要素逐级分解,并对其进行难度系数打分(难度系数一至五星,难度依次增加)从而得出无菌控制难点,分析如下:
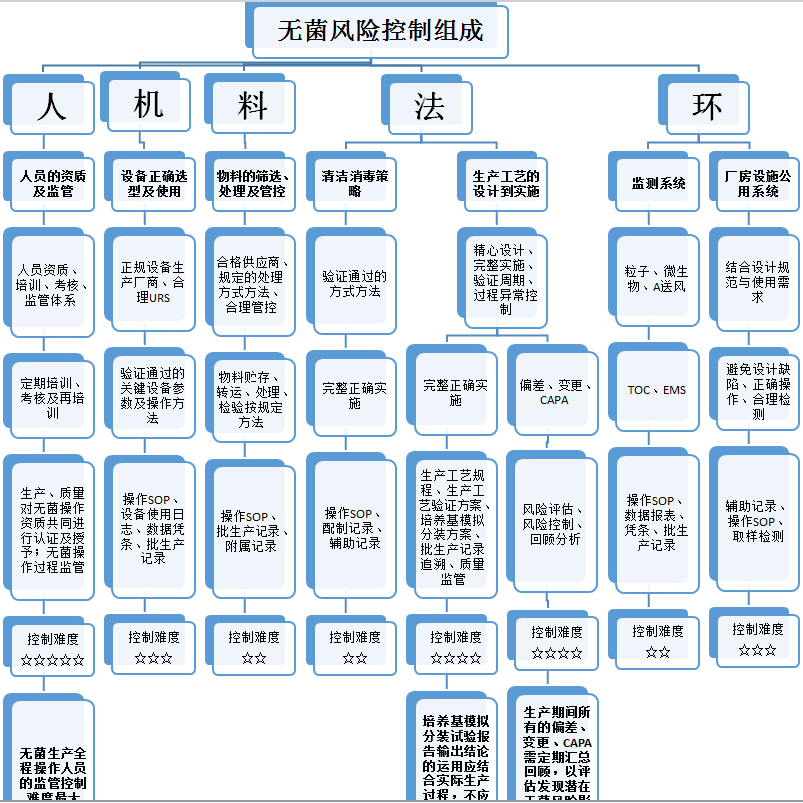
从上图分析可明显看出,难点一共有三个,其中:
难度四星的有两项,分别为无菌生产过程出现的偏差、变更、CAPA及培养基模拟试验输出结论的正确运用;
难度五星的有一项,为无菌生产过程操作人员的监管控制。
难点一
无菌生产过程出现的偏差、变更、CAPA
偏差、变更、CAPA作为药品质量保证要素的“三架马车”,也是我们常用必不可少的工具,其重要性此处不再赘述。药品生产过程出现并体现偏差、变更、CAPA是很正常的事,尤其是偏差会经常发生,无菌生产过程不发生偏差才是极不正常的现象。关键是发生了偏差该如何制定纠正预防措施(包括应急措施)将风险降至最低或可控,争取同样问题下次不会重复出现。
当然,此处要声明一点,并不是说药品生产过程出现偏差越多越好;例如,生产10批次,生产周期也就一个月,但期间出现了10个左右偏差,其中还有相同偏差;像这种情况,有可能是某项甚至几项生产要素压根就不具备稳定生产的条件,像这种情况可能就需要停产发起CAPA或变更给予纠正解决了。
一个好的药企,年底在做质量回顾时,应该是变更多才对,而不是偏差满天飞;因为不管是生产体系还是质量体系贵就贵在持续改进完善,而变更就是其体现的载体。
为何要把无菌生产过程出现的偏差、变更、CAPA列为一个无菌控制难点,很多人心里都会觉得这些要素谁没见过,谁没写过,难在哪里?其实,把它列出来是想体现一个理念,那就是:无菌生产出现的偏差、变更、CAPA一定不要仅留意于表面,写完关闭就完事了,其实阶段性的回顾、统计、分析才是重点,这会让你发现、发掘潜在的无菌风险影响因素,让你在无菌控制上始终保持一定的前瞻性,从而做到提前预知、提前准备,消除潜在无菌风险影响因素。
难点二
培养基模拟试验输出结论的正确运用
无菌工艺模拟试验(培养基模拟试验)是指采用微生物培养基替代产品(无菌粉末分装的培养基灌装试验实际是将产品替代品溶入培养基中,不是直接替代产品)对无菌工艺进行评估的验证技术。
精心设计并完整实施该模拟试验,目的就是检验无菌生产工艺整个无菌保障体系的有效性。培养基模拟试验的“灵魂”就是“最差条件的模拟”;结合最差条件精心设计方案去挑战并得出可实际参考并运用的结论来指导无菌生产。例如:无菌保持时限、非正常干预操作等都是由培养基模拟试验挑战得来。
之所以将培养基模拟试验输出结论的正确运用列为无菌控制难点,依旧是要强调一个理念,那就是:因为培养基模拟试验与实际无菌生产终归存在差异,所以不能将培养基模拟试验得出的一些结论与实际生产中的无菌污染风险等同;实际生产过程整体无菌风险的评估是大前提,是基础,在这个基础上引用培养基模拟试验输出的结论才可行。
举个粗俗易懂的例子:一条粉针灌装线培养基模拟试验得出的灌装时限为14小时,结合日常生产经验,文件规定的灌装时限为10小时,有一批次生产因为灌装设备异常频发,导致在文件规定的时限10小时内未灌装结束,此时该企业直接引用了培养基挑战时限14小时,将灌装时限延长了4小时灌装结束。像这种情况就属于片面引用培养基模拟试验输出结论;因为培养基模拟试验时未出现与该批次生产过程一样的异常,在引用时,未充分评估该批次实际生产过程出现的无菌风险。
难点三
无菌生产过程操作人员的监管控制
无菌工艺生产过程,最大的污染源就是“人”,人员操作导致污染发生的可能性超过70%;生产人员的技能、所接受的培训及其工作态度是无菌操作合规的关键因素。人员进入关键操作区域的人数、人员资质确认、人员操作技能培训及考核,这些工作多下功夫,车间管理人员及QA负责任的话相信不难办到。难点就是操作人员的“工作态度”,因为无菌生产操作人员也是人,是有情绪、想法的活体,如何保证他们每次操作的重现性、合规性及每步重要操作都能受控,这就很难。
无菌操作人员可能存在的风险
①手套频繁接触设备表面、工作台面、墙面或维修设备结束后未更换新的无菌手套、手消,仍从事核心A层流下无菌工作;
②更衣不规范,无菌衣被污染穿入或身体部位未有效全部遮挡进入核心区域从事无菌操作;
③在核心高风险区域运动、动作幅度过大破坏洁净区气流流形,带起大量尘埃粒子;
④无菌操作不专注,导致出现不必要的偏差(在未密封瓶口上方直接操作)等。
上述提到的风险都是无菌操作人员无菌操作的细节,这些细节全部都受控,不漏一丝的监管很难达成;但也不是没有一点办法,我们除了日常监控外还可以制定一系列其他措施,以便更好的服务、监管无菌操作人员,让“工作态度”这个因素“受控”,以降低无菌控制难度。
①合格、良好的无菌操作相关内容培训、资质确认及绩效考核——清楚自身职责的重要性及合理对等的考核。
②企业需重视无菌操作人员的薪资待遇——对自身工作有积极性。
③负责任的班组长很重要,视情况给予其一定的授权——对无菌操作细节监督和指导。
④无菌操作人员梯队搭建——资质传递、健康竞争评比环境。
⑤制定人员无菌操作监控计划——包括但不限于:人员上岗前身体、心理状况确认;无菌操作细节抽查;手套、洁净服取样。
上述对无菌控制的三个难点进行了分析并得出结论,其实更多的是理念性的东西。对于从事无菌药品生产的人员来讲,无菌理念(亦可理解为无菌意识)的重要性不亚于无菌操作技能;意识先行,技能跟上,无菌控制也没有多难,当然这都要建立在无菌药品生产企业重视无菌生产的基础上。
参考资料:
《药品GMP指南—无菌药品》
《无菌药品生产发生微生物污染的因素分析》
关于举办中药饮片炮制技术研究与质量控
各会员单位: 为顺应药品监管规..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


