

迄今为止,全球已发现6000至8000种罕见病,而且这个数字还在增加,每年有近250种新疾病被列为罕见病。
如何治疗罕见病,如何发现治疗罕见病的新药,是全球制药业面临的共同问题。
目前,全球只有10%的罕见病具有相应的疗法,有4000多种罕见病完全没有相应的治疗药物。
因此,通过对目前罕见病的治疗方法和治疗药物,包括已经上市的药物、最新的临床试验和正在研究的热点药物分析和总结,可以为从事罕见病研究的人员提供参考。
不过,罕见病的准确诊断和及时治疗是临床医生面临的共同挑战。遗传缺陷是造成罕见病的一个重要因素。遗传性罕见病的治疗方法包括饮食疗法、手术、药物治疗、骨髓移植等。小分子药物疗法、抗体疗法、酶替代疗法、基因疗法、干细胞疗法和药物重新定位是用于治疗罕见病的一些方式。特别是寡核苷酸疗法、干细胞疗法和基因疗法等新的研究热点的出现,改变了许多复杂疾病的治疗模式,为罕见病患者带来了新希望。
基因疗法
基因疗法是一种通过改变人类基因来治疗和预防疾病的技术。腺病毒载体是基因疗法中最常用的病毒载体之一。基因疗法不仅能够治疗血液病,如血友病、戈谢病和血友病,而且在治疗遗传病、罕见病和肿瘤方面也取得了重大进展。
遗传是罕见病的一个关键因素,大多数罕见病是由单一基因的突变引起的,这些基因可以从父母那里遗传,也可以在生殖细胞中自发发生。
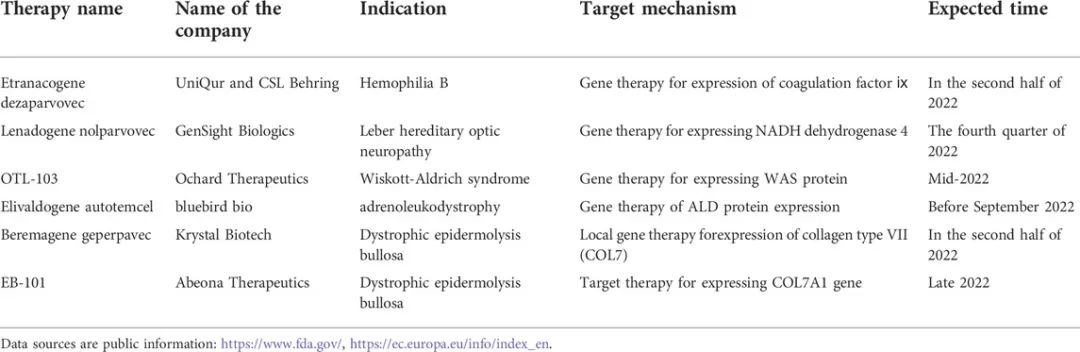
大多数基因疗法被用于治疗罕见疾病。Glybera(alipogene tiparvovec)是欧洲药品管理局(EMA)在2012年批准的第一个基因疗法药物,由荷兰公司UniQure开发,用于治疗超级罕见疾病脂蛋白脂肪酶缺乏症。从那时起,基于DNA的基因疗法已经在罕见疾病领域取得了一些突破。欧美获批基因疗法见表1。此外,根据公开信息,一些基因疗法有望在2022年被批准上市或提交BLA,其中可能在罕见病领域获得进展的基因疗法见表2。
表1.批准上市罕见疾病基因药物[1]
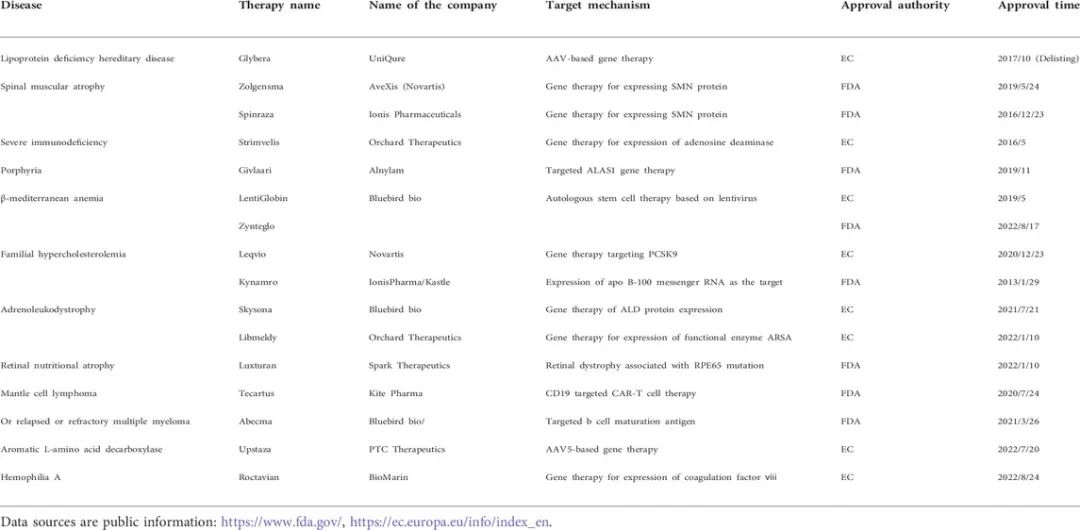
表2. 预计2022年将在罕见病领域获得批准的基因疗法清单[1]
干细胞疗法
造血干细胞移植(HSCT)是一种使用人类细胞来调节免疫系统或提供护理支持治疗方式。
HSCT的移植途径包括胎儿干细胞移植、骨髓移植、外周血干细胞移植和脐带血移植。许多临床实践和应用表明,脐带血在治疗罕见疾病方面具有良好的效果。以往的研究表明,脐带血干细胞(UCBT)在治疗原发性罕见病方面有明显的疗效,但免疫重建缓慢和巨细胞病毒(CMV)感染率高是干细胞疗法不可忽视的缺陷。
尽管如此,脐带血在治疗罕见病方面还是具有不错的前景。例如,脐带血给免疫缺陷综合征、变色性白血病、慢性肉芽肿病、IgE综合征等罕见病带来了重生的希望,在多种溶酶体储存病(粘多糖储存酶、戈谢病、尼曼病等)中也取得了良好的治疗效果。
戈谢病是最常见的溶酶体储存疾病,由葡萄糖脑苷脂酶缺乏引起,是一种常染色体隐性疾病。目前这种疾病的治疗方法包括酶替代疗法(ERT)和造血干细胞移植,这种方法可以为戈谢病患者提供永久性的酶源,而且与常用的干预措施ERT相比,造血干细胞移植的费用更低,早在1984年就已经移植治疗戈谢病。
默沙东公司的Prevymi(letermovir)口服液和静脉注射液,有助于阻止造血干细胞移植后的CMV感染,于2017年获得FDA批准。Prevymi是FDA批准的第一个治疗CMV感染的新药,在促进移植的同时也加强了感染的预防和控制,提高了造血干细胞移植的成功率。造血干细胞移植正逐渐被认可和接受,在罕见疾病中有着广泛的应用。
抗体疗法
抗体疗法是一种被动免疫,即人体在体外被动地接受抗体,从而获得特异性免疫力。抗体通过调节信号转导途径发挥作用,将细胞或蛋白收集到特定部位,然后传递细胞毒性或中和循环因子。
Cablivi(caplacizumab)是第一个可用于治疗获得性血栓性血小板减少性紫癜成年患者的纳米抗体药物,于2018年被欧盟委员会(EC)批准上市。
抗体疗法的开发主要是针对患者人数较多的适应症,然后转移到一些罕见病的适应症。例如,Alexion Pharm公司的Soliris(依库珠单抗)是一种针对末端补体蛋白C5的单克隆抗体,于2007年首次被FDA批准用于治疗阵发性夜间血红蛋白尿。此外,Soliris被批准用于治疗另外两种与补体系统密切相关的罕见疾病:非典型溶血性尿毒症和重症肌无力。
总之,抗体疗法的一个主要优势是高特异性,减少了小分子制剂中常见的脱靶毒性的风险,对治疗罕见疾病至关重要。
酶替代疗法
酶替代疗法(ERT)可用于治疗部分溶酶体储存疾病(LSD)。第一次利用ERT治疗LSD是在1980年,Brady及其同事创造了一种用于1型戈谢病的酶替代疗法。这成为随后治疗LSD的原则性证明。
ERT是一种生物疗法,最初于1991年被批准用于戈谢病。健赞公司开发了一种重组形式的葡萄糖脑苷酶,1994年首次被FDA批准。为治疗戈谢病、法布雷氏病、庞贝氏病和粘多糖储存病,已有15种酶替代疗法罕见病药物被批准在国际上销售。
表3 国外已上市罕见病酶替代疗法[2]

迟发性庞贝氏病(LOPD)是一种罕见的疾病。自2006年以来,ERT已被批准用于治疗庞贝病。研究表明,ERT可以改善迟发性庞贝氏病患者的运动呼吸功能。Nexviazyme(艾夫糖苷酶α)是一种酶替代疗法,于2021年被FDA批准用于治疗1岁及以上的迟发性庞贝氏病患者。
可以看出,ERT发展的重点一直是各种LSD,作为一种遗传性疾病,引起溶酶体的酶缺乏或酶错误。在ERT治疗疾病多年的基础上,ERT治疗具有技术发展较好、安全性较高的优点。但是,也存在生物药物在体内分布不明确、难以穿越血脑屏障、治疗费用昂贵等问题。为了成功地进行ERT的临床试验,关键是要确定相关的研究终点,并认识到这些终点的最小临床意义的病人差异。
参考资料:
[1] Han Q, Fu H, Chu X, Wen R, Zhang M, You T, Fu P, Qin J, Cui T. Research advances in treatment methods and drug development for rare diseases. Front Pharmacol. 2022 Oct 12;13:971541. doi: 10.3389/fphar.2022.971541. PMID: 36313320; PMCID: PMC9597619.
[2] 程速远,寇雅真,罗建辉. 罕见病酶替代疗法药物研究进展及药学评价思考[J]. 中国药学杂志,2020,55(13):1128-1132. DOI:10.11669/cpj.2020.13.014.
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


