

“一个人一辈子做出来一个创新药就非常了不起了,我见证了这个家伙(彭彬)做出来的两个(创新药)。”
前昆泰大中华区总裁、昆翎医药联合创始人张丹习惯在公开场合总是这样提起彭彬。而他说这番话的背景是,创新药研发的基本面本就是一场九死一生的冒险,临床失败率高达95%。
彭彬现在是一家成立仅5年的中国创新药公司的首席医学官,但他也是在肿瘤创新药临床转化和试验领域超过二十年的专家——他在世界级药企诺华工作多年,是划时代抗癌靶向药格列卫的临床试验药理团队负责人。
这款被《我不是药神》传播的家喻户晓的药,是人类历史上第一个根据致癌基因设计药物,开启了抗癌药“特异攻击”癌细胞的先河,原本患绝症的病人,通过这款药,可以像高血压、糖尿病患者一样带病生存。
它的诞生开辟了癌症靶向治疗的新时代,也成为此后资本涌入创新药领域最吸引人的故事。
1998年刚做完博士后的彭彬被派往诺华总部,负责一款名为“CGP57148”的临床试验。那时,即便是诺华总部,对这款药也不看好。
耗费数亿美元但针对的疾病“慢性粒细胞白血病”在美国只有几千患者,诺华为是否开发这款药,此前已犹豫了2年多——除了商业的考虑外,成药性也很难预知,需要彭彬这样的药理学家解决无数个难题,才能落地。
此前,诺华前身公司Ciba刚结束了一个不大成熟的药品的研发,而那个年代开启了核酸药的一波研发高潮(现在大热的mRNA疫苗即属此类)。当时,大批公司扎堆研发核酸药,却因为无法解决核酸药突破人类细胞膜的问题,全部失败。
但没想到到是,诺华无意中推进的这款“CGP57148”,因临床试验效果特别好而迅速通过FDA审批,以“格列卫”的商品名为人熟知。
幸运的不只诺华,还有负责格列卫临床药理团队的彭彬,这成为他“一辈子都值得骄傲”的事情。
十几年后,彭彬以诺华全球转化中国区负责人的身份推动了另一个创新药成功上市。那时是2011年前后,他也开创了跨国药企在中国进行早期临床项目的先河。
随后几年,彭彬见证了毕井泉上任后,一系列中国药审领域的改革,目睹中国创新药井喷式的发展——中国在创新药领域,和世界最发达国家的距离越来越小。
在科研领域,第一代抗癌靶向药惊艳亮相之后,癌症这个众病之王随之狡猾抵抗、不断升级,患者对靶向药的耐药性出现,抗癌药也需要一次次升级换代。作为药理学家的彭彬依旧在研究新的靶点,探索千万分之一的成药可能。
最终,担任外企高管多年的彭彬,索性自己也投入了中国创新药领域,成为一家研发双抗抗癌药创新药企的首席医学官。目前双抗也是抗癌药物领域最前沿的道路之一。
令人意外的是,格列卫诞生时代一度大热又被打入冷宫的“核酸药物”,终于解决了如何进入人类细胞膜的问题。mRNA疫苗在新冠疫情中脱颖而出,这一领域再次成为热门赛道。
时代的螺旋上升,并非场景重演,而是制药业的特点。
药品的研发,因为一个理论产生想象空间,又因为一个难题停止,在解决困境中不断向前。成功者,除了才华、努力和热情之外,往往多了那么一点运气,还有赌对时代的一点勇气。
彭彬的临床药物研发经历,更像是一部浓缩的创新药简史,让我们得以窥见新药研发的振奋光影,以及挫败和灰暗时刻。
-01-
“格列卫问世后,我每年都关注诺贝尔奖”
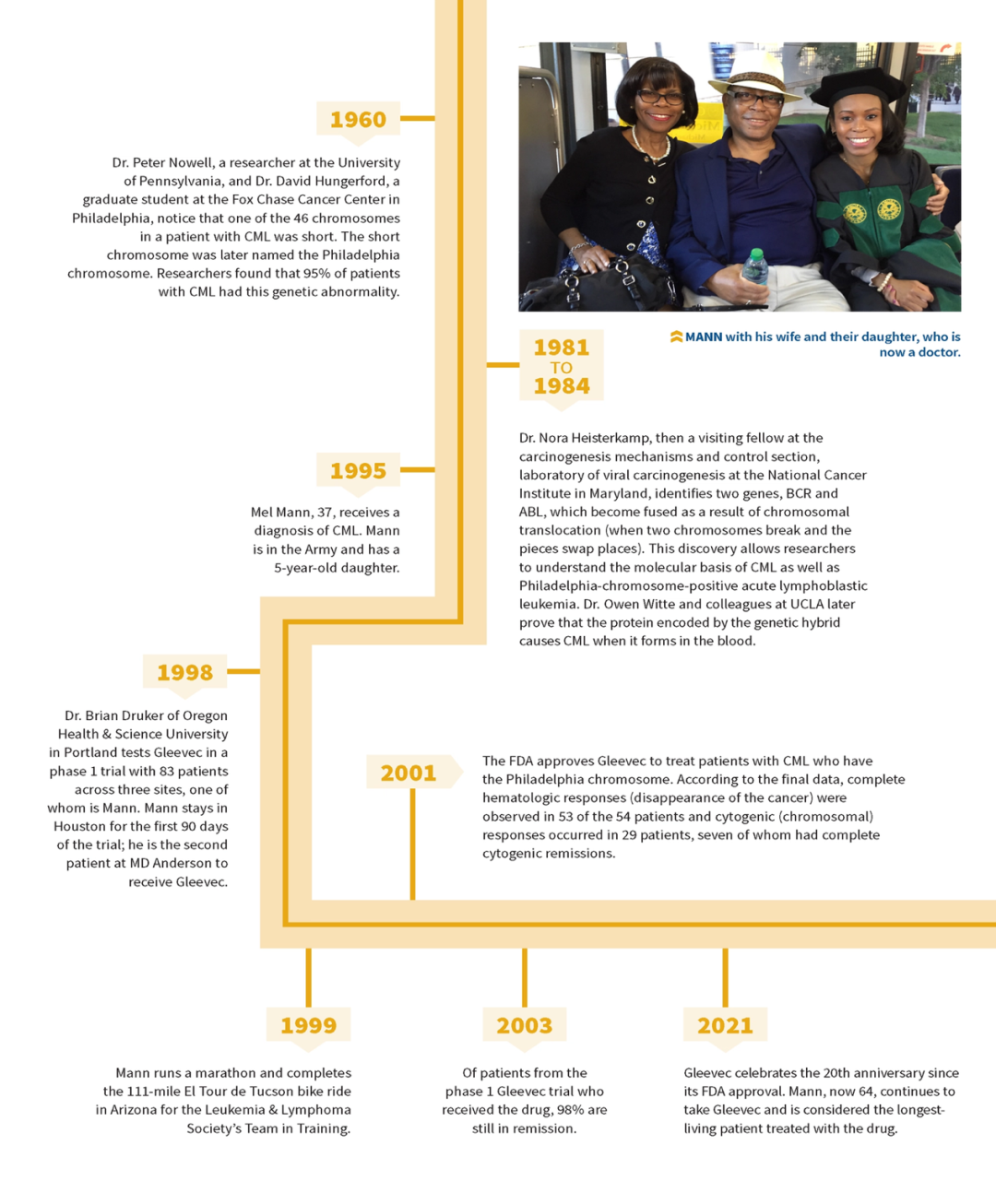
对于64岁的Mann来说,他37岁以后的人生是橘色小药丸从死神手里抢来的。
他是目前接受格列卫治疗的最长寿的生存者。当时37岁的他可能不曾想过,自己能健康活到女儿的大学毕业典礼。
27年前,Mann曾被确诊患有慢性粒细胞白血病(CML),该疾病也是格列卫的主要治疗领域。当时医生面对这样的患者,常常告诉他们:这是重病,病程致命预后很差,平均生存期可能3-6年。
而现在医生会告诉病人,这是一种病程缓慢的白血病,只要终生口服格列卫,通常都能颐养天年。
“神药”并非虚言,CML患者的预后从5年生存率30%增加到了10年生存率85%~90%。且在5年后,依旧有98%的患者取得了血液学上的完全缓解。
换句话说,它改写了癌症治疗的面貌:虽不能治愈癌症,但却能把一种绝症转变为像高血压、糖尿病一样可控可治的慢性病,患者可以带癌长期生存。
现在肿瘤学家讨论CML这一致命疾病时,会使用“前格列卫时代”和“后格列卫时代”这种说法。实际上,格列卫出现的意义不仅限于某个病种,它更为人类的抗癌血泪史画上了一个革命性的注脚。
在与癌症鏖战的大半个世纪中,外科手术、化疗和放疗一直是最主要的三种癌症治疗方式。它们轮番上阵,却都有着诟病已久的局限。
比如外科手术对于适用范围和癌症早晚期的效果差异大,其癌细胞的残存几率也不小;而化疗和放疗则是“地毯式轰炸”,好的细胞和坏的细胞同时都被破坏,多数患者要承受脱发、恶心呕吐等副作用的折磨。
作为全球第一个靶向药物,格列卫换了种战略,它像经过严格训练的“猎癌狙击手”,直接瞄准癌细胞或者它最脆弱的环节,枪枪中的而不会“滥杀”正常细胞,大幅降低副作用并提高效果。
自此格列卫为癌症治疗推开了一扇新的门,这也标志着癌症分子靶向治疗的时代来临了。2001年5月,美国食品药品管理局(FDA)鉴于格列卫惊人的临床试验数据,仅10周的审查时间就核准其上市,用于治疗CML。这也创下了当时FDA历史上最快的一次药物审批。
随后它被美国《时代》周刊称为“银色的子弹”,被ASCO评选为癌症治疗50年五大进展之一。
另外,2009年格列卫的主要开发者布莱恩·德鲁克尔、尼古拉斯·莱登和查尔斯·索耶斯获得了被誉为“诺奖风向标”的美国“拉斯克奖”。遗憾的是,直到2021年也没有听到格列卫的研发者获得诺贝尔奖的消息。
“自从格列卫问世以后,我每年都关注诺贝尔奖。”格列卫临床药理试验的负责人彭彬这样告诉深蓝观。
-02-
“神药”格列卫:从赔钱药到卖出40亿美元
实际上,“神药”差点被放弃。
在 I 期临床试验开始前,很多人并不看好格列卫甚至拿它当做赔钱药。背后的原因主要有两个,一是它针对的CML患者人群比较小,美国每年仅有几千名患者受CML折磨。
1996年诺华正式成立,它由原瑞士汽巴嘉基集团(Ciba-Geigy,年营收约200亿美元)和山德士集团(Sandoz,年营收约60亿美元)两个集团对等合并而来。后期预计将耗费1-2亿美元进行动物试验和临床试验,但花费上亿元只能造福数千人,这让诺华犹豫不决。“变成这么大的体量却要target这样的小众人群,性价比不是很高。”彭彬认为这是高层的考量。
对于诺华而言,该药精确的专一性正是其致命的有限性——投入很多,受众很少,注定是赔本买卖。
另外一个原因是,那时还没有任何一款靶向药研发成功的案例。“当时业界比较热衷的是核酸药,但很少有突破。”彭彬解释当时因为两个拦路虎——药物分子跨膜递送和自身免疫,当时全世界研发的核酸药几乎全军覆没。
那时患了CML,唯一的也是首选的治疗方法是骨髓移植。但合适的配型非常难找,仅有20%至25%的患者拥有移植的资格。其次在接受移植后,患者还总伴随严重、甚至致命的副作用。
1998年,Mann也被医生告知骨髓移植是其延长寿命的最佳机会。就在他艰难地寻找合适的骨髓配型时,一位病友向他介绍了安德森癌症中心即将进行的格列卫 I 期临床试验。
终于,诺华被格列卫的开发者说服,同意合成并发放差不多刚好给约100名患者试用的CGP57148(格列卫的前身,后更名为STI571)。
研发者可以放手一搏了,但机会只有一次。
后来Mann和一群经过治疗但病情依旧严重的患者,每日口服格列卫,正是参与了这次前途未卜又雄心勃勃的临床试验。
回忆起领导格列卫临床 I 期试验的日子,彭彬表示是自己的运气。其博士后师从国际临床药动学鼻祖、英国曼彻斯特大学药学系Malcolm Rowland教授,刚读完加入了诺华在瑞士的总部。
他还记得主管当时这样说:“首先你是新兵,另外这个药物的市场潜力可能没那么高。”综合了两方面的因素,主管决意让其作为I期临床试验临床药理团队的负责人。
在 I 期临床试验中,研究人员先后爬坡了25毫克到150毫克的几个剂量,却没有看到病人有任何反应。
转机发生在剂量为200毫克的时候。“三个病人在口服格列卫一个星期后,体内的白细胞全部降到正常范围之内。”而当剂量跃升为300毫克以后时,发生了堪称奇迹的疗效:在54名患者中有53名出现了血液学上的完全缓解(CHR)。
CML主要两个重要的诊断指标:白细胞飙升和费城染色体阳性(大约95%的CML病例都是阳性)。彭彬表示,在剂量为200毫克时,除了所有病人的白细胞降到正常,在骨髓穿刺检查中,70%-80%患者的费城染色体都转为阴性。
火箭终于成功升空。
在彭彬及其团队成功研究出格列卫兼顾有效性和安全性的最佳临床剂量后,他以第一作者或参与作者的身份在国际著名杂志发表了一系列格列卫的临床研究论文。
“我真的很好奇,这个药怎么能做(骨髓)手术呢?” 当时医生告诉Mann,这是一种靶向药物,它只会针对“坏细胞”而不影响“好细胞”。
所谓的“坏细胞”应该指的是骨髓造血干细胞的基因突变,即产生了费城染色体,而此染色体会形成Bcr-Abl融合基因(酪氨酸激酶TKI),后者激活了迫使细胞不断分裂的通路。
既然治不了本,那就通过抑制酪氨酸激酶来控制CML的增殖。“就有点像高血压糖尿病人,也许永远不能根治,但是至少保证白细胞维持在正常水平。”彭彬解释了格列卫的药物作用原理。
而Bcr-Abl之所以能够成为施药的完美目标,还在于它具有“口袋”状的凹陷。而靶向治疗简单来讲就是锁匠配钥匙,改变钥匙的形状然后试着开锁,如果不合适就再次打磨。
“它真的是我的救命稻草。”Mann想起当时的情景,不禁感慨格列卫是神药。而参与临床试验的医生们也很兴奋,认为诺华找到了一个真正好的药。“对我来言也学了一课——做临床研究还是要有耐心。”彭彬说道。
那是1999年,正值因特网聊天室风行,可喜的药物临床数据在网络上迅速传开。在短短三周的时间内,3000多人集体寄信给诺华总裁,目的是想让诺华扩大试验规模,让格列卫能早日上市。诺华决定呼应这个请求,随即展开了 II 期临床试验。
在该项临床试验中,97%的病人用药6周后白血球数量恢复正常,75%的病人体内费城染色体细胞消失。更令人惊喜的是,这些疗效相当持久:在治疗的一年半后,患者的无进展生存率依然达到了89.2%。这种纪录前无来者。
彭彬犹记得在2001年,自己被代表诺华团队在ASCO(美国临床肿瘤学会)年会上首次报告格列卫的 I/II 期临床药理实验结果,ASCO是目前全球最大最具影响力的肿瘤组织。
当时会场上有上万人,也是从那个时候开始肿瘤界的人开始认识他。“当时,整个会议上也就一两个像我这样的亚裔上台发表报告。”
21年前,一个鲜少的华人面孔说着带有口音的英文,在肿瘤界最高规格的学术大会上发表了重量级的报告,这也是华人在新药研发领域的标志性事件。此后,彭彬还分别参加了另外两个TOP级别的大会——ASH(美国血液学会年会)和AACR(美国癌症研究学会)。
2001年5月,基于 II 期临床试验的积极疗效,格列卫获得了美国 FDA(食品药物管理局)加速审批,用于干扰素治疗失败的急变期、加速期或慢性期 Ph+ (阳性)CML患者治疗。
从 I 期临床试验到最后上市,格列卫只用了三年的时间,这也是当时FDA审评史上最快的药物。据统计,新药开发平均耗时10-15 年时间。其中临床试验是耗时最长的阶段,I 期至 III 期平均耗时 6-7 年。加速审批并非意味着一劳永逸,格列卫在上市后补充了III期临床试验,效果完胜标准疗法。
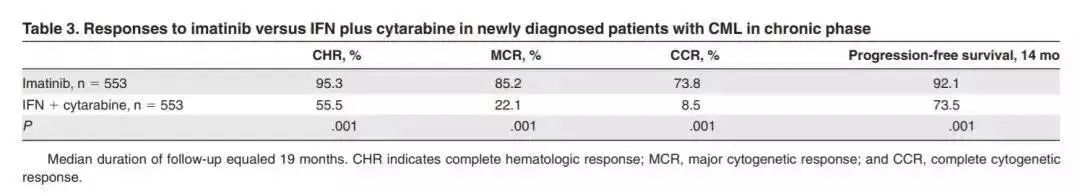
△格列卫III期临床试验数据对比
上市后,格列卫的销售额逐年攀升,三年即突破10亿美元,这也是诺华首个破10亿的“重磅炸弹”。此后其销售成绩继续增长,在2011年达到巅峰46亿美元。
彭彬坦言:“我们当初做临床试验的时候,没有意识到会有现在这么大的市场。”他分析这主要得益于格列卫将CML这种病逆转为“慢性病”后,伴随着新患者的增加,旧的患者凭借吃药也获得了长期生存。
“神药”亦有瑕疵。其实在其上市后不久,就有一些患者出现了耐药性,癌症卷土重来。
针对这个问题,在CML治疗领域,BCR-ABL酪氨酸激酶抑制剂经历了三代更迭:从第一代伊马替尼(格列卫)到第二代尼洛替尼(达希纳)、达沙替尼(施达赛)以及新型二代氟马替尼,现在正在向第三代迈进,代表性的药物包括普纳替尼和耐克替尼(亚盛医药)等。
△格列卫研发历程
图片来源:Cure
-03-
跨国药企首次在国内开展早期临床试验
2011年,彭彬被派往上海组建诺华在中国的肿瘤转化研究中心。和上次格列卫的临床试验不同,这次他带着高选择性口服小分子MET抑制剂capmatinib(Tabrecta®)来到中国,想要在国内以及亚太地区开展全球早期临床项目,比如 I 期临床试验。
彼此,国内在MRCT(国际多中心临床试验)方面的法规仍然延续2002年颁布的《药品注册管理办法》(试行)。其中明确规定:临床研究用药物应当是已在境外注册的药品或已进入临床 II/III 临床试验的药物。
那个年代还比较忌讳外国药企到中国做药物的 I 期临床试验,盛行着“拿中国患者做小白鼠的论调。彭彬首先就遇到了一个大难关,“我们做首次人体临床试验的IND都递不进去。”
彭彬没有就此止步,他想到了一个妥协的办法。既然法规要求药物需要进入 II/III 临床试验,那么就等韩国、新加坡等国家结束了 I 期临床试验,再在中国开展 II 临床试验。
他在递交的材料里写明这点后,辗转递交给CDE(国家食品药品监督管理局药品审评中心)。CDE的专家看到材料的说明确实不违规,最后成功接收了。
随后戏剧性的一幕发生了,专家的审评意见用白纸黑字写着:希望诺华不只在中国做 II 期, I 期爬坡临床试验也要参加,我们也需要中国人的 I 期临床数据。
峰回路转,最后反而把事情办成了。诺华算是赶上了政策过渡期的好时候,喝到了外企在中国做早期临床试验的“头啖汤”,这让彭彬在诺华再次成为“明星”。
那时彭彬察觉出,中国的相关法规正在酝酿着改变,CDE的专家也认识到了旧法规的不合理。一个客观的趋势是,当时制药外企在国内临床研究的数量快速增长。
CDE的数据显示,2005年前,制药外企在选择中国开展MRCT上数量较少,基本只是观望和尝试,而2005 -2011年它们在这方面的临床试验数量较之前显著上升。
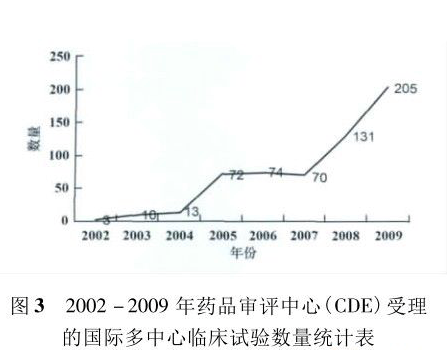
图片来源:
《中国药物临床试验发展面临的机遇与挑战及政策建议》
此后CDE还多次召集了多家制药外企的高管,针对MRCT早期试验沟通和交流。彭彬就曾参加过,“我们和CDE表达的意思是,允许外企做早期临床试验其实能带动国内企业。”
伴随《国务院关于改革药品医疗器械审评审批制度的意见》(下称《意见》)在2015年发布,其明确规定“允许境外未上市新药经批准后在境内同步开展临床试验”。这也意味着CDE不再限制国外药企在中国开展MRCT的 I 期临床试验。
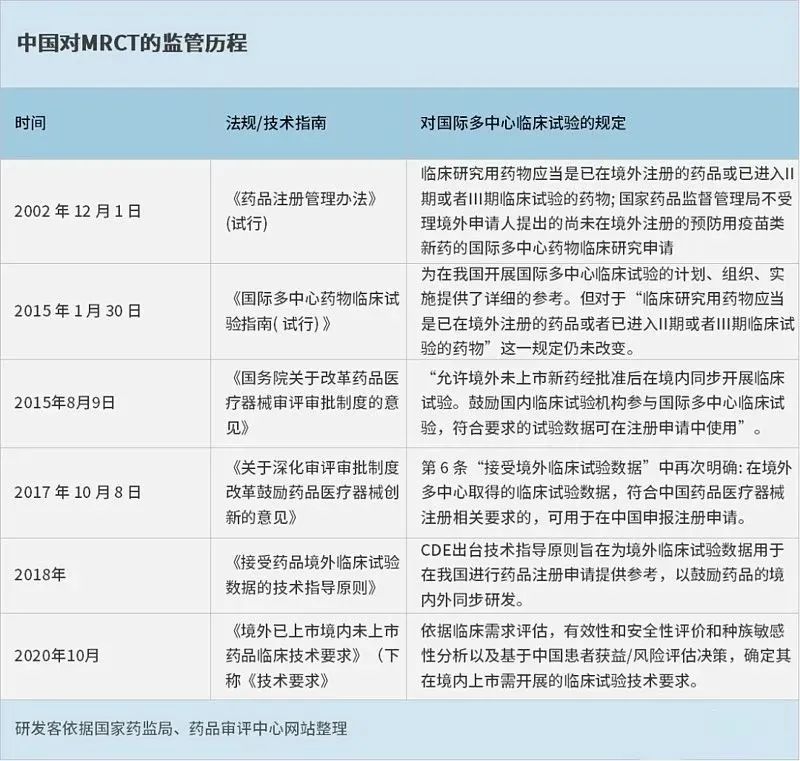
△中国对MRCT的监管历程
图片来源:研发客
此外,2017年中国加入ICH(国际人用药品注册技术协调会),进一步助推了医药的国际化接轨,药物审评现代化走向快车道。
2019年新《药品管理法》正式颁布,其鼓励境外原研药品在临床早期研发阶段在中国开展同步研发,如以MRCT的方式。
过去的几十年,国内药企更多地生活在药监局的庇荫下。只靠仿制药供血的药企前途黯然,中国制药产业得不到发展是法规改革的深刻背景。
像《意见》、《药品管理法》等政策的出现,放进来了制药外企“这条鲶鱼”,搅活国内制药业的创新潜力。一定程度上,它们也迫使中国制药业走出舒适区,和全球高水平竞争。
默克雪兰诺全球研发中心中国区临床开发中心负责人史冬梅,曾在接受媒体采访时表示:“跨国公司将创新产品带到中国,让中国顶尖研究者参加早期临床试验,从引入GCP(药物临床试验质量管理规范)理念到参与政策法规的制定都发挥了重要作用。”
政策的支持催化了MRCT的繁荣,药物研发日渐全球化,也让MRCT日益受到重视。MRCT的好处不仅可以加快新药同步研发,试验结果能用于多个监管机构注册审评,而且可以优化患者资源和节约研发费用。
近五年来,在中国开展的首次人体试验显著增加,从2016年的363项增长至2020年(1至9月数据)的975项。目前,跨地区和国家的监管机构逐渐接受 MRCT数据作为支持药物(医药产品)批准上市的主要证据。
-04-
奔赴下一代创新药
临床试验的大门打开了只是第一步,漫长的研发期才刚刚开始,这也是新药研发的常态。
诺华的这款小分子MET抑制剂capmatinib(Tabrecta®)在2011年进入临床 I 期试验,但直到去年它才在美国获批上市,被视作肺癌靶向药耐药后的新希望。
当初作为该药的临床试验带头人,彭彬早在它上市前的2018年就离开了诺华。如果除去2005-2009年在GSK美国的4年,他在瑞士、美国以及上海三地为诺华工作了16年。
2018年,国内一家创新药企业——岸迈生物的CEO吴辰冰博士,刚刚完成了一个双抗的临床前研究。这个双抗靶向肿瘤细胞的EGFR和cMET。
那时这家企业在临床试验方面欠缺经验,吴辰冰通过多方打听了解到彭彬在诺华专门做cMET靶点的研究。“他来找我喝咖啡,咨询怎么做cMET的临床(试验)的问题,我给了他一些参考建议。”聊过几次后,有一天吴辰冰直接向他发出了“入伙”邀请:“不知道你感不感兴趣,我们一起来做(创业)。”
在制药巨头工作工作了二十年后,彭彬开始对国内的biotech动心了。
彼时国内的创新药领域方兴未艾,国内药企正从单纯的模仿/改进(Me-too/Me-better)到快速跟进(Fast-follow)甚至逐步追求First-in-class。
药政审评审批制度改革提速,带量采购模式出现,这些都将国内药企引向创新的潮头。除了政策红利,资本市场也开始助力。2018年港交所主板上市规则新增第18A章新规,允许未有收入、未有利润的生物科技公司提交上市申请。
这家公司研发的药物靶点他非常熟悉,他日夜研究cMET的通路,很看好这个双抗成药。
双抗也是彭彬想要做出下一个milestone(新药研发的关键节点)的药物研发领域。双抗即双特异抗体,指的是可以同时结合两个不同抗原,或者是一个抗原不同表位的抗体。它如同一块“磁石”,将T细胞引导到癌细胞附近,使其定向杀伤肿瘤细胞。因此它被称为“第二代”免疫治疗药物,与单抗相比,其在疗效提升、副作用降低等方面具有优势。
截至目前,全球已有4款双抗药物上市,其中两款已在国内获批。而国内已有60余款双特异性抗体进入临床阶段,涉及恒瑞医药、信达生物、百济神州、康方生物、康宁杰瑞等三十多家企业。其中进度最快的是康方生物的AK104 和康宁杰瑞的 KN046,均已进入 III 期临床。
而在2018年,国内研发双抗的药企还没有几家。
像之前的所有创新药一样,双抗的研发会面临实践中的困境,许多世界级药企也面临极大失败的可能。靶点的选择和药物本身的成药性是关乎双抗研发成败的关键。而研发失败除了本身药物实力“不够硬”,还可能是操作层面上的经验不足导致的。
罗氏一个cMET单抗的失败案例,让彭彬反复复盘,“II期试验数据非常漂亮,III期试验却失败。可能主要是 III 期临床选择的特定人群出错了。”
另外一个相似的案例是治疗非小细胞肺癌的EGFR抑制剂吉非替尼,吉非替尼的临床 II 期结果也非常好,但到了 III 期结果显示欧美失败,但中国和亚太地区却成功了,这是因为这两个地区的人群EGFR靶点的突变率较高。
在国外制药巨头积累了几十年临床研发经验,回望过去10年,彭彬没有想到中国如今已经有非常好的临床研究。在他眼里无论是临床研究的数量还是质量,国内外已经没有太多区别。
上次彭彬参加ASCO的年会是2019年,距离他代表诺华参加这场世界肿瘤领域最负盛名的年会已经过去了18年。
2001年,ASCO年会出现一个中国人做报告是极其罕见的事情,但在2019年的ASCO年会上,世界顶级肿瘤研究专家公开表示,全世界做CART-T临床研究最多的已经不是美国而是中国了。
“下一次ASCO年会召开的时候,或许就有人该说,中国开展的双抗临床研究数量也已成为世界之最。”而彭彬,或许也会等来他的第三次幸运。
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


