

近日,复旦大学、西安交大、中国医学科学院等26家单位联合发布了中国人群泛基因组联盟(CPC)一期研究进展。6月14日,相关成果以《基于36个族群的中国人泛基因组参考图谱》(A Pangenome Reference of 36 Chinese populations)为题发表于《自然》(Nature)主刊。

这项研究初步构建了中国人群的泛基因组参考图谱,发现了在人类通用参考基因组上缺失的约1.9亿个碱基对的参考序列;新鉴定了约580万个点突变或小变异以及3.4万个结构变异;并发现通用参考基因组上缺失的参考序列富集了适应性演化和起源于远古人类的遗传变异,并且与角质化、紫外线辐射应激、DNA 修复、免疫反应以及寿命等表型或功能相关。在重构人类演化历程、挽回复杂疾病研究时“丢失的遗传率”等研究和应用中,该图谱具有巨大的潜在价值。

▲“中国人群泛基因组联盟”一期36个族群画像集(图片来源:研究团队供图)
人类参考基因组是广泛用于人类遗传学和医学研究的遗传密码“导航图”,也是解析人类起源与演化、解析人类表型和疾病的遗传基础的根基。从2001年首次发表人类基因组草图,人类参考基因组经历了数十次的更新迭代。直到2022年 “端粒到端粒”联盟(T2T)构建的“无缺口”的T2T-CHM13参考基因组完成图,所有涉及人类遗传学的研究仍然依赖于线性参考基因组。
今年正值国际人类基因组计划(HGP)完成20周年,人类参考基因组从“线性一维序列”过渡到“泛基因组多维图谱”。泛基因组(Pangenome)进一步借助图论的思想和计算技术,将人类多个族群的代表性样本的具有多样性的基因序列以多维图谱形式组装起来,形成一个能充分反映种群基因组结构变异多样性的导航图,从而指导进一步的遗传学和医学研究。
在人类基因组计划成立之初,主要的人类参考基因组皆基于高加索人种为主体样本构建,难以代表其他族群的基因组多样性。作为世界人口大国,中国有着丰富的人群多样性。构建具有代表性的中国人群泛基因组图谱,将极大提高捕获罕见或低频遗传变异的灵敏度和准确性,有望应用于复杂疾病的分子机制研究,以及精准医学研究。
为了构建高质量高精度的中国人群泛基因组参考图谱,复旦大学徐书华教授、西安交通大学叶凯教授联合中国26家单位发起了中国人群泛基因组联盟(Chinese Pangenome Consortium, CPC)。
在其第一期研究计划中,CPC对代表中国36个族群的58个样本采用最新的第三代高保真基因组测序技术进行了深度测序,结合最新的单倍型基因组组装方法,获取了116个高质量单倍型基因组,并以图基因组的方式构建了高质量中国人群参考泛基因组。该泛基因组图谱总共包含约3.01 Gb个碱基对的序列信息,在现有人类参考基因组的基础上新增了约1.9亿个碱基对的新序列,包含约590万个小变异(单核苷酸多态性变异和小规模插入/缺失变异)和约3.4万个结构变异(Structural variation, SV),涉及至少1367个蛋白编码基因复制事件等。其中,约500万个碱基对新序列存在于95%以上的单倍型中,被视为中国人群基因组核心序列,可能与中国人群特有的较为稳定的生物学功能或表型特征相关。
CPC泛基因组图谱作为首个中国人群专属的泛基因组参考图谱,与HPRC泛基因组图谱相比,在中国人群特有的复杂变异解析方面具有显著优势。CPC泛基因组图谱中新发现了1079个基因拷贝数变异,以及包含药物代谢基因CYP2D6等在内的在中国人群中富集而在其他世界人群中出现频率较低的若干基因拷贝数变异;新鉴定出富集在中心粒、端粒等染色体复杂区域的3.4万个结构变异,其中半数以上仅在单个或两个样本中出现——若不针对中国丰富的族群多样性开展专门研究,将没有机会发现这些遗传变异。
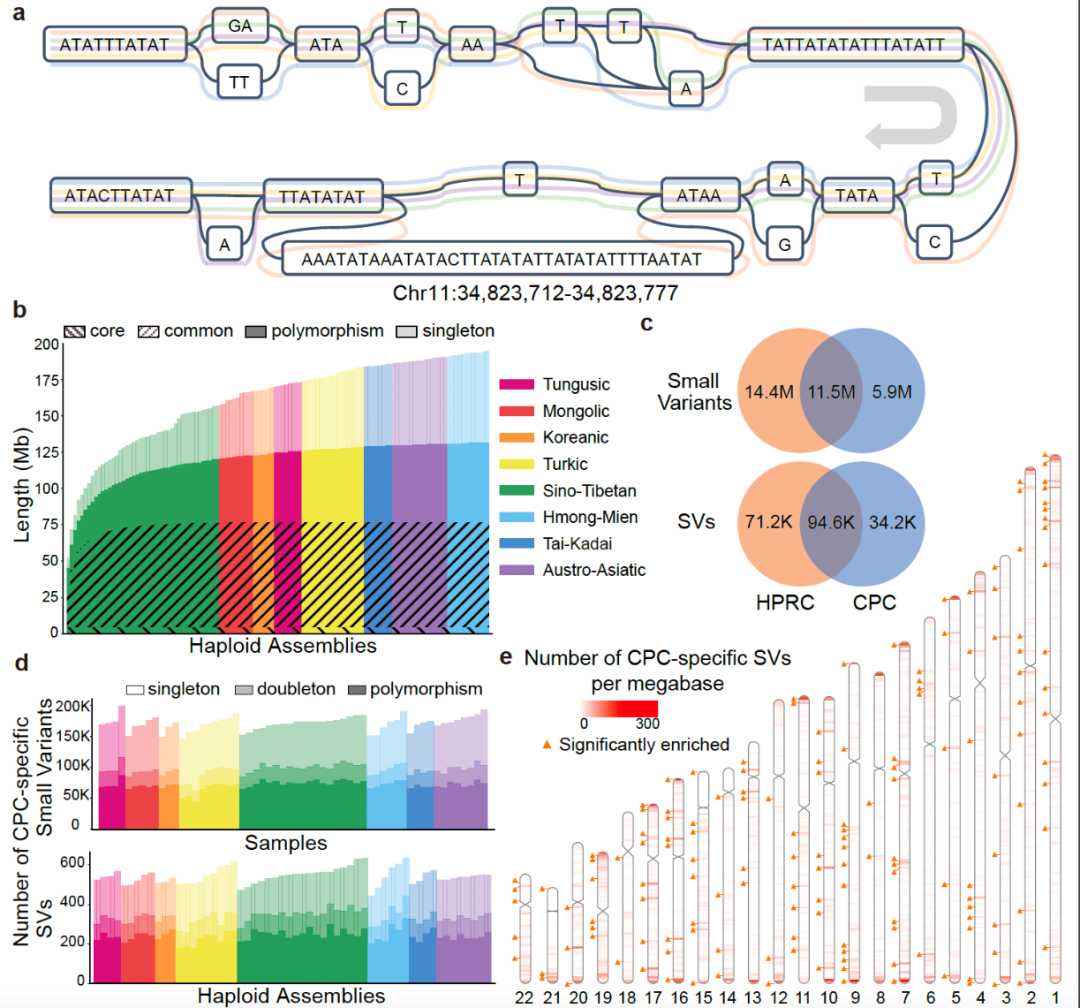
▲图形化泛基因组示例及CPC特有复杂变异分布(图片来源:参考资料[1])
研究人员进一步揭示,这些CPC新发现的遗传变异可能与亚洲人群特有的疾病易感性及表型多样性有关。一个典型的例子是α-珠蛋白基因簇,研究人员在该基因区域鉴定出两个中国人群特异性的大规模结构变异,包括一段20 kb的缺失序列和一段10 kb的重复序列,这将为进一步研究中国人群贫血症的遗传机理和致病机制提供新的线索。
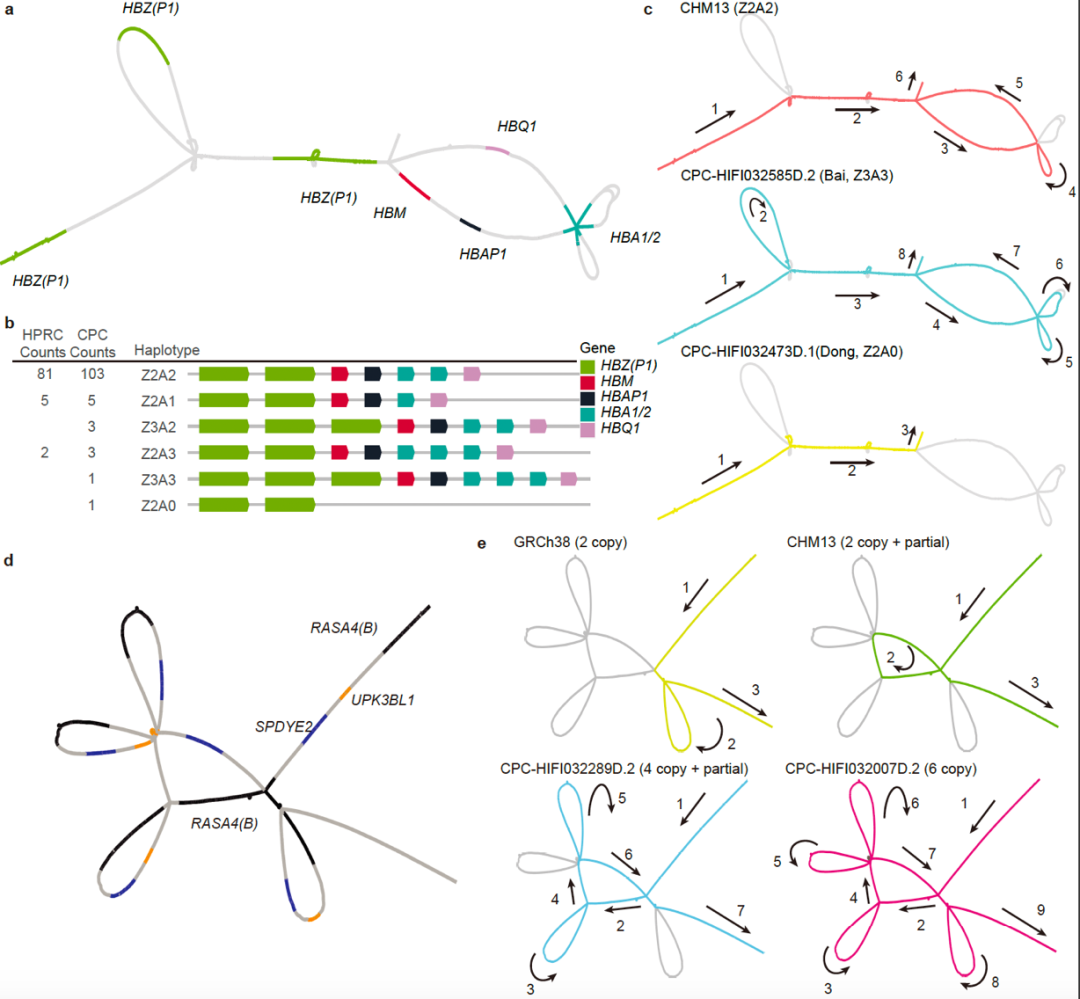
▲CPC对中国族群特异的复杂基因组结构变异解析示例(图片来源:参考资料[1])
同时,CPC新发现的遗传变异影响了具有潜在功能和经受过适应性进化的基因,这些基因可能与亚洲人群特有的疾病易感性及表型多样性有关,这也证实了将人群专属高质量泛基因组用于基因组学和医学研究的潜力和必要性。此外,研究人员在CPC参考图谱中发现了相当大比例的古人来源基因序列——平均每个族群和每个样本中分别有约15 Mb和约9.5 Mb的古人来源新序列——这可能是前期开展大量研究却未在现代人基因组中发现的古人基因渗入序列,或将为东亚现代人基因组中的古人基因渗入研究乃至整个古DNA领域提供新的信息资料和线索。
从人类基因组计划中国只承担“1%”的图谱绘制任务到今天中国人群泛基因组图谱“100%”由中国科学家完成,这项研究成果展现了中国生命科学尤其是基因组学科研水平在过去40年间的历史性跨越,为完整构建中国人群参考泛基因组打下了坚实的基础,也为人类参考泛基因组图谱绘制了“中国画卷”。中国人群参考泛基因组不仅有助于加速遗传学研究,加深人们对个体或群体基因组的“异”与“同”更具象、更深度的认知,还将改变过去依赖主体基于高加索人种的参考基因组而导致东亚特有罕见变异检出精确度难以提升的困境,从而提高中国生物医学数据分析的质量和效率,服务人民生命健康。
复旦大学徐书华教授、西安交通大学叶凯教授、中国医学科学院褚嘉祐教授和复旦大学陆艳副教授为论文的共同通讯作者。复旦大学博士后高扬,西安交通大学杨晓飞副教授,中国科学院上海营养与健康研究所博士生陈豪、谭昕江,中国医学科学院杨昭庆研究员以及复旦大学邓恋青年研究员为论文的并列第一作者。该论文共同作者包括复旦大学王宝楠、孔爽、李松洋、崔雨杭、雷畅、赵晓涵、史颖冰、杨子艺,中科院王亦民博士、潘雨闻博士、马森博士,中国医科院孙浩博士,中科院昆明动物所吴东东研究员,江苏师范大学伍少远教授,复旦大学赵兴明教授,西安交大附属医院施秉银教授,复旦大学金力教授,南京医科大学胡志斌教授等。
中国人群泛基因组图谱已公开在线发布:
https://pog.fudan.edu.cn/cpc/#/
https://github.com/Shuhua-Group/Chinese-Pangenome-Consortium-Phase-I
该项研究所涉及的样本信息和数据的公开发表已获得国家人类遗传资源管理部门批准。该项研究得到了国家自然科学基金重点项目、基础科学中心、国家重点研发计划等项目的资助。
附:中国人泛基因组联盟(1期)成员
徐书华(复旦大学)、叶凯(西安交通大学)、褚嘉祐(中国医学科学院)、金力(复旦大学)、杨昭庆(中国医学科学院)、吴东东(中国科学院昆明动物研究所)、胡志斌(南京医科大学)、赵兴明(复旦大学)、袁慧军(四川大学华西医院)、高强(复旦大学附属中山医院)、金鑫(华大生命科学研究院)、陈超(中南大学)、李津臣(中南大学)、杨剑(西湖大学)、伍少远(江苏师范大学)、陆艳(复旦大学)、沈侠(复旦大学)、杨晓飞(西安交通大学)、邓恋(复旦大学)、樊少华(复旦大学)、戴俊程(南京医科大学)、施秉银(西安交通大学)、韦朝春(上海交通大学)、孙浩(中国医学科学院)、何光林(四川大学华西医院)、卜枫啸(四川大学华西医院)、吴洋(四川大学华西医院)、高扬(复旦大学)、陈豪(中国科学院上海营养与健康研究所)、谭昕江(中国科学院上海营养与健康研究所)、王亦民(中国科学院上海营养与健康研究所)、王宝楠(复旦大学)、李松洋(复旦大学)、崔雨杭(复旦大学)、潘雨闻(中国科学院上海营养与健康研究所)、马森(中国科学院上海营养与健康研究所)、孔爽(复旦大学)、赵晓涵(复旦大学)、毛创学(中国科学院上海营养与健康研究所)、雷畅(复旦大学)、史颖冰(复旦大学)、杨子艺(复旦大学)
专家点评
赵国屏(中国科学院院士)
由复旦大学、西安交大、中国医学科学院等研究单位的一批中青年学者组织的“中国人泛基因组联盟”,汇聚中国各方面多年努力积累的数据,构建出高质量的中国人群泛基因组图谱,在国际顶级科学期刊Nature正式发表,是一件值得庆贺和重视的大事。
人类基因组计划(HGP)最主要的成果就是构建了一个沿用至今并在不断完善中的人类参考基因组(reference genome)。人类参考基因组之所以重要和有用,因为人类虽然是一个物种,其基因组大致相似(如果不考虑结构变异,在组成人类基因组的30亿个碱基对中,大约99.9%是一致的),然而,人类个体的基因组又是各不相同的;而正是这0.1%左右(大约三百万碱基对)的不同,造成了人类各族群、个体间表型不同(从长相的不同到健康状况的差异)的遗传基础。所以,要能够正确使用人类基因组序列,帮助我们认识人类自身特别是与我们健康密切相关的基因组信息,以获得有价值的知识,必须有一张能够指导待研究对象的基因排序和组合的导航图,这就是人类参考基因组。
这张被广泛应用于人类遗传学和医学研究的遗传密码“导航图”,是解析人类起源与演化、解析人类表型和疾病的遗传基础的根基。20年前人类基因组计划完成之初,用于国际基因组计划(HGP)的大部分DNA来自于住在纽约州布法罗的一名白人男性捐献者(编号为RP11)。同时公布的来自Craig Venter领导的塞雷拉基因組计划的DNA样品来源于五名捐献者,其中虽然有亚裔和非洲裔人,依然是高加索人为主。因此,通用的人类参考基因组代表的是以高加索人为主的基因组序列。
今年正值国际人类基因组计划(HGP)完成20周年,也成为人类参考基因组从“线性一维序列”过渡到“泛基因组多维图谱”的转折年。值此历史发展重要转折点,复旦大学、西安交大、中国医学科学院等研究单位的一批中青年学者,能独立自主地构建出高质量的中国人群泛基因组图谱,最终得到国际顶级期刊的认可和发表,这是值得肯定的,也是需要鼓励和支持的。这一成果表明中国科学家在人类基因组学领域的研究水平得到了显著提升。我相信这项工作对中国的人类基因组学和医学中的复杂疾病遗传基础研究等领域会起到重要的推动作用!
张亚平(中国科学院院士)
基因组是生命演化的遗传信息载体,也是研究人类起源和复杂疾病的数据信息根源。不论是动植物,还是人类,复杂性状的遗传学研究都遇到了一个瓶颈——“missing heritability”(丢失的遗传率)问题,即目前的研究未能鉴别出能充分解释表型预期遗传贡献的基因变异。这部分与参考基因组的不完整性和多样性覆盖度不充分有关。虽然国际上也发起了以欧美发达国家主导的重构人类参考基因组的计划,然而,是否有必要建立中国人群的参考基因组是应该认真对待的问题。
徐书华团队在前期研究中从变异判读、进化研究、医学应用三个方面分析和评估了建立高质量族群特异性参考基因组的必要性。此次复旦大学、西安交大、中国医学科学院等多家单位联合发布的中国人泛基因组研究成果,再次表明了建立中国人群泛基因组图谱的必要性以及该图谱在重构人类演化历程和挽回复杂疾病研究中“丢失的遗传率”等研究和应用中的潜在价值。
金力(中国科学院院士)
人类基因组研究是基因组学中起步最早的方向,在数据资源、技术方法和研究成果等方面都曾领先于其他物种的研究。但是,这种“先发优势”也使得人类基因组研究越来越难以取得新的突破性成果。近年来,动物、植物、非人灵长类、反刍动物等领域的基因组学研究进展很快,取得了令人瞩目的成绩。相比之下,人类基因组研究显得有所“滞后”。而此次发表的成果,打破了人类基因组学研究的这种“停滞”,也是中国科学家在人类基因组研究领域取得的又一重大进展。
很有意思的是,这项研究发现通用参考基因组上缺失的参考序列富集了适应性演化和起源于远古人类的遗传变异,并且与角质化、紫外线辐射应激、DNA 修复、免疫反应以及寿命等表型或功能相关。这反映了我们的基因组中可能仍有大片有价值的矿藏尚未得到充分开采。
值得注意的是,这项研究除了在通用参考基因组之外发现约590万个单核苷酸多态性变异和插入/缺失变异之外,还鉴别出约3.4万个结构变异(Structural variation, SV),并且涉及至少1367个蛋白编码基因。我想强调的是,基因组结构变异大概是生物进化中从微观到宏观演变的关键遗传基础,也是最有可能连接渐变到跃变这个“鸿沟”的进化密码。我相信,通过对基因组结构变异的高精度解析,不但能大幅提升“基因型-表型”关联分析的功效,而且有可能最终帮助我们理解生命演化中重要性状和功能产生的遗传基础和分子机制。
参考资料:
[1] Gao, Y., Yang, X., Chen, H. et al. A pangenome reference of 36 Chinese populations. Nature (2023). https://doi.org/10.1038/s41586-023-06173-7
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


