

在本周五(5月12日)对Sarepta Therapeutics公司杜氏肌营养不良症(DMD)基因疗法SRP-9001召开专家咨询委员会会议之前,美国食品药品监督管理局(FDA)发布了简报文件,对其安全有效治疗患者的能力表示担忧。
FDA在审查了所有证据后指出,迄今为止进行的临床研究并没有提供明确的证据,证明SRP-9001可能对DMD的门诊患者有益。
“从申请人提供的数据中合理确定地得出SRP-9001可能对年轻患者有效,或者对年长患者或功能状况较差的患者无效的结论,这是一项挑战。”
该机构还指出了与SRP-9001相关的安全问题“与实施无效基因疗法的可能性有关”。
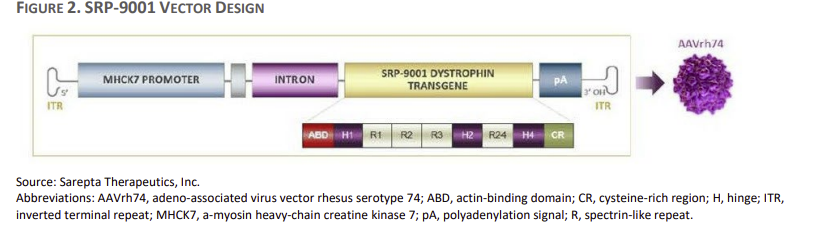
细胞、组织和基因治疗咨询委员会召开会议,讨论四个关键议题。然而,只有一个首要问题将被投票表决:考虑到不确定性,现有证据是否支持加快批准SRP-9001,使用替代终点治疗确诊DMD基因突变的DMD患者?
问题1:替代终点是否有可预测临床效益,并获得加速批准
Sarepta提出的替代终点是否有可能预测临床效益,该终点将使SRP-9001有资格获得加速批准。
基于该基因疗法在第12周刺激微肌营养不良蛋白表达的能力,Sarepta公司已要求提前批准。
FDA怀疑是否有任何证据表明这种生物标志物证明了SRP-901的药理作用,但它只是表明该蛋白已在靶细胞中表达。
SRP-901旨在将DMD的疾病改变为一种较温和的形式,称为贝克肌营养不良症(BMD)。为了做到这一点,据说基因疗法可以纠正肌肉中肌营养不良蛋白的表达,Sarepta认为这将纠正DMD的根本原因。
早在2018年12月,FDA就对SRP-9001的临床益处表示担忧,当时该机构对替代终点提出质疑,并建议Sarepta根据文件中显示的监管时间表,选择一个“评估临床意义益处的终点,如患者的感觉、功能或存活情况”。该问题在2020年9月、2021年7月和2022年4月与该公司的各种会议上再次被提及。
Sarepta在提交生物制品许可申请(BLA)前的最后一次互动中,引用了监管先例,允许加速批准促进肌营养不良蛋白缩短形式表达的药物。FDA反驳说,这不一定等同于预测临床益处以支持加速批准。
BLA于2022年9月28日提交。
Sarepta在其提交的材料中辩称,通过自然史研究表明,蛋白的替代具有临床益处,并减缓疾病进展。
Sarepta说:“长期以来,DMD患者恢复功能性肌营养不良蛋白一直被认为是治疗DMD的一个关键且被广泛接受的治疗目标。”Sarepta认为SRP-9001已经被证明可以做到这一点。该公司提出了多项不同的临床前研究来支持这一点。
问题2:仅有唯一一项临床研究数据可用
第二个问题涉及SRP-9001的唯一一项随机、双盲、安慰剂对照临床研究,该研究是迄今为止可用的数据,即2期阶段研究102的第一部分。主要目标是评估治疗对北极星动态评估(NSAA)的影响,这是一种常用的评定量表,用于评估DMD患儿的功能性运动能力、进展和治疗效果。与安慰剂相比,该研究在第48周没有显示出这一指标的统计学显著变化。接受基因治疗的组与基线相比没有任何改善。
相反,Sarepta指出了一项亚组分析,该分析表明4至5岁的门诊患者的预后更好。但6至7岁的患者没有任何改善。当时,该公司将其归咎于试验的两个部分之间的明显不平衡,认为安慰剂患者在基线时对非甾体抗炎药的功能评分更高。
根据FDA的说法,Sarepta声称,接受SRP-901治疗的患者的NSAA评分与基线相比的平均变化“在所有时间点都更大”。
该机构写道:“FDA的评估是,SRP-901组和安慰剂组在所有时间点的差异都在不确定性范围内,甚至缺乏统计显著性的趋势也证明了这一点。”。
后来,FDA表示,虽然年龄是DMD疾病进展的一个重要因素,但事后分析并没有预先指定。
FDA表示:“在整个人群中进行总体无显著性测试后的特设亚组测试只能被视为假设,因此必须谨慎解释这一亚组分析。”。
Sarepta提供了SRP-901整个临床项目的证据,以支持临床疗效,包括研究103、102和101,并对试验进行了综合分析。研究103是一个正在进行的开放标签1期,这意味着患者知道他们正在接受基因治疗,NSAA评分评估是一个探索性目标。Sarepta说,第1组患者在第52周时NSAA评分平均增加,一年后评分显著增加。
研究101——一项开放标签、单臂1/2期测试——同样显示分数增加,并持续在四年。
至于研究102,Sarepta再次指出了NSAA的基线失衡,并表示结果“难以解释”
Sarepta解释道:“三项试验的治疗效果和NSAA总分的总体模式是一致的、持久的,可归因于SRP-9001。”。
问题3:风险不确定性
FDA已要求委员会在第三个问题上考虑SRP-9001基因治疗在门诊患者中的风险。
FDA正在标记AAV基因治疗类别的总体安全性,该类别与治疗突发的严重不良事件有关,包括肝毒性、器官损伤、贫血甚至死亡。一些动物研究也显示了癌症的潜在可能性,尽管人类肿瘤的形成尚未得到证实。
FDA表示,在SRP-901的风险收益分析中必须考虑这一类风险。
根据FDA的统计,“85名受试者共发生1230起治疗突发不良事件”,但研究中没有观察到死亡。11名患者出现严重不良事件,最常见的报告问题是呕吐、恶心、急性肝损伤、发烧和血小板低。
Sarepta注意到了类效应,并表示SRP-901治疗在第4周至第8周期间观察到了肝损伤,但这些问题已经解决,没有出现并发症。该公司表示,这些事件是可以预测的。
Sarepta注意到DMD的罕见性,他说这三项研究代表了“182.75患者年的暴露时间”,并证明“SRP-9001总体上是安全的,耐受性良好。”
SRP-9001在2017年10月开始的开发过程中被搁置了两次临床试验。2018年6月,FDA首次暂停了该项目,原因是人类患者“已经或可能面临不合理的重大疾病或伤害风险”,而人体检测申请没有足够的风险评估信息。三个月后,临床搁置被解除。
第二次搁置发生在2021年8月,当时FDA再次指出缺乏风险评估信息。与此同时,据报道,一名9岁患者在服用SRP-9001后需要住院治疗和呼吸支持,出现严重不良事件。
问题4:如上市能否完成验证性研究
上周,Sarepta高管在第一季度电话财报会议会议上预告了他们可能会面临的问题。首席执行官Doug Ingram预测了临床益处的讨论,但也表示FDA可能对Sarepta能否完成一项验证性研究存在疑问。
FDA确实要求委员会审查这项验证性研究,Sarepta正在通过3期阶段301研究的第一部分进行这项研究,该研究被称为EMBARK。但该机构更具体地要求委员会考虑早期市场准入可能对完成该试验产生的影响。
该研究对120名4至8岁的患者进行了全面的研究,并以52周的随机双盲形式对SRP-901与安慰剂进行了对比测试。还有一个名为第二部分的交叉部分将于2023年6月开始。
Sarepta表示,一旦SRP-9001进入市场,患者退出的风险“极低”,因为美国以外的试验中有45名患者,这将是唯一可以使用基因疗法的地方。
瑞穗证券(Mizuho Securities)分析师Uy Ear表示,简报文件“可能会使咨询委员会会议难以进行积极的投票”。
FDA不必遵循咨询委员会的投票,而是通过讨论来做出决定。
Ear在周三给客户的一份报告中写道:“总的来说,我们认为FDA的简报读起来非常不好,FDA基本上指出,目前可用的数据不支持加速审批。”
但投资者显然认为单一投票问题是一个积极的迹象,因为FDA没有要求委员会考虑是否应该等到EMBARK完成后再批准。
投资者仍然认为FDA可能在5月29日之前批准该药物。
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


