

孤儿药美国将罕见疾病定义为影响该国20万人以下的疾病,或“没有合理预期”收回研发成本的疾病。
近几十年来,FDA已经批准了数百种罕见疾病新药(孤儿药),但对于影响美国人和全世界数百万人的7000种罕见疾病,仍然需要大量的新疗法。
鉴于对药物开发的固有风险和罕见病治疗所能获得的收入极小患者群体的关注,多年来,政府采取了各种激励措施,鼓励制造商开发罕见疾病药物。1983年的《孤儿药品法》(ODA)预示着这些努力的开始,该法案为孤儿药的临床试验提供了更长的市场独占性和税收减免。后来出台的其他政策和计划也通过为孤儿药开发提供费用减免和研究资助来支持孤儿药的开发。
几乎没有异议的是,官方发展援助和额外的好处促进了获批治疗罕见病的产品数量,1983年至2019年期间有超过800种孤儿药适应症获得FDA批准。这一进展也受到了细胞和基因疗法等科学发现的刺激,这些科学发现彻底改变了对许多罕见疾病的治疗标准。
这种成功不应掩盖一个事实,即绝大多数的罕见病仍然没有得到获批上市的治疗药物,这意味着大量的需求没有得到满足。
但是,开发更多孤儿药以满足这一需求的希望,现在被日益严重的可负担性问题所笼罩。
2019年,在美国每个接受治疗的病人平均每年的孤儿药治疗费用为32,000美元,治疗费用从每年6000美元到500,000美元不等。大多数孤儿药的上市价格很高,39%的孤儿药每年的费用超过10万美元,基因和细胞疗法的费用为数十万美元或更高。对于每年定价10万美元的药物来说,接受治疗的患者群体只有1万人,每年产生的收入确达到了10亿美元——即孤儿药"重磅炸弹"。
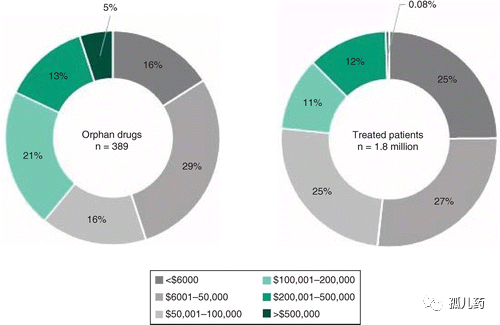
图1. 2019年"孤儿药"和其适应症药物治疗的患者之年度药物成本
因此,今天,随着美国FDA每年新批准的获得孤儿药的比例达到50%以上,孤儿药有关的主要挑战不再是创造一个可行的商业模式,而是在越来越多获批上市的高价孤儿药的费用方面,面临着越来越大的挑战,这可能会威胁到可持续的保险费水平,并为个体患者的使用设置更大的障碍。对支持FDA批准孤儿药的证据质量的担忧放大了这种负担能力的挑战。虽然高药价是整个治疗领域的一个问题,但由于孤儿药依赖于使用短期替代结果的非随机试验,因此通常在相对安全性和有效性方面的证据较为有限的情况下被批准。
尽管有其他有助于促进孤儿药发展的风向标,包括科学进步、新的FDA审批途径和有限的竞争,但孤儿药的溢价仍持续存在。科学进步使治疗方法更精确地针对潜在的疾病机制,使孤儿药申请的成功率更高,降低了生命科学公司和投资者的风险。
其次,FDA加速审批途径的建立,简化了许多符合该审批途径的罕见病产品的证据要求。
最后,不足的潜在利润和预期的市场规模可能不会吸引仿制药竞争者进入该市场。
是否还需要目前水平的溢价作为激励措施来推动孤儿药的开发,目前还存在激烈的争议,但关于孤儿药审批的数据表明,科学进步、监管灵活性、市场条件和溢价能力的结合,使孤儿药成为对投资者和生命科学公司有吸引力的市场。
因此,孤儿药的未来问题包括以下信号:
创新正在蓬勃发展,但尚未达到满足未满足临床需求的水平;
官方发展援助所建立的激励机制的基础设施在推动创新方面至关重要,但现在可能被推动对罕见疾病的投资和创新的其他因素所掩盖;
越来越多孤儿药的成功并没有引发价格下降,因此正在造成财政压力,有可能破坏这些治疗的获得和所有患者的医疗保险的可负担性。
医疗系统的所有参与者,包括患者、创新者和支付者,都会同意,目标应该是建立一个政策和实践的基础设施,在患者和医疗系统都能负担得起的平台内推动创新。
对于我们这些追随者来说,尽管目前广大罕见疾病患者尚无药可用,不过同样需要思考:在政策和实践中是否有正确的平衡来实现这一目标?以及未来如何避免重走美国的路?
备注:随着近年来定价屡创新高孤儿药基因疗法获批上市,将会进一步拉升患者平均治疗费用。
参考资料:
[1] Pearson C, Schapiro L, Pearson SD. The next generation of rare disease drug policy: ensuring both innovation and affordability. J Comp Eff Res. 2022 Oct;11(14):999-1010. doi: 10.2217/cer-2022-0120. Epub 2022 Aug 10. PMID: 35946484.
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


