

3月14日/15日,在美国北贝塞斯达举行的ISPE无菌会议上,PIC/S主席PaulA.Gustafson概述了由PIC/S、WHO和欧盟合作修订的欧盟GMP指南附录1的修订进程:该文件处于最终通过阶段。预计将在今年7月初至9月底之间发布。欧盟委员会目前正在等待PIC/S和WHO的审查。
针对本次修订的欧盟无菌技术规范附录,并没有出现“跨越发展条目修订”,而是结合目前技术发展所做出的部分修整与进一步解释细节而已。
无菌产品质量属性实现与持续保证,其核心事项在不同国家、地区、组织所颁布的技术文件(指南)中强调与表述不尽相同,主要核心要点总是形影不离万变不离其宗,即:
1.制剂类别与微生物限度控制需求及决策
2.微生物控制方法与技术决策及最终产品无菌状态保证
3.货架期间无菌状态与持续无菌贮藏状态保证
4.最终实现无菌产品工艺属性状态与概述
5.工艺与设备、厂房选择与决策一致性
6.产品生产技术实现与洁净级别一致性
7.过程微生物控制能力管理原则
8.过程无菌状态与硬件持续维护
9.状态无菌保障能力设定与一般数据证明分析确认管理
10.取样代表性与无菌状态持续保证一致性管理
11.无菌状态破坏及影响因素确证结果对应风险评估与决策
12.最低质量成本与无菌状态品保证
13.无菌产品过程控制相关人员培养与持续监测
14.质量回顾与质量趋势事件涉及微生物异常的调查
上述关注内容在不同技术指南文件具体描述要求,以及配套验证管理指南类技术文件表述与要求。
更新集中在4点内容
针对本次修订的欧盟无菌技术规范附录,并没有出现“跨越发展条目修订”,而是结合目前技术发展所做出的部分修整与进一步解释细节而已。
在总则范围继续阐述“风险管理为主要技术评价原则”,并未出现“颠覆性”内容改变。
如果说有改变,也是基于“具体风险管理实践技术要求”同步更新而已。新的内容集中在:
1、再次拓延“风险管理习惯与意识”,在具体风险管理技术框架基础上要结合“历史数据、经验、患者影响数据”综合评价。
2、硬件选择与使用,突出“技术手段实现人员限制禁入高风险区域”,而不是仅仅通过“在线监测技术数据分析方式”去证明“人员进入限制区域”风险可接受。
3、污染控制策略(CCS)应在无菌产品的“厂房设施决策全过程实践并有足够客观监测加以证明”,通行的“风险评价可以接受一些可接受低风险习惯监管策略”已经不再被认同,至少需要逐渐抛弃。
4、附录中所列具体“最低硬件和范例区域划分案例”,一方面可以理解为“符合即通过”;另一方面则是提出“仅作为判定原则阶段共识”,应随着地区水平差异性、技术发展科学性进一步“讨论、纳入、列为行业共识”管理。
提升“管理科学性”思路,重新梳理国内技术规范
总的来说,国内习惯的“指南要求就是一定符合、指南给出描述就是基本共识”的思路需要改一改了,这些提升的“管理科学性”思路,已经在国内《疫苗生产检验电子化记录技术指南(征求意见稿)》中窥见一斑。
摘选内容:
(内容导读请在下列文本框中上下滑动查看)
本指南制定了在疫苗生产过程中,与疫苗生产、检验过程电子化记录相关的业务要求和技术要求。
本指南同样适用于疫苗生产和检验过程质量管理信息化相关系统的设计原则和评审依据。
对于本指南未列举的信息化相关业务数据,疫苗上市许可持有人可参考本指南的相关内容,基于风险并按照同等原则执行电子记录。
电子批记录的生产部分至少要包含以下内容:
[1]产品名称、产品编码、规格或批量和批号的电子数据
[2]生产以及中间工序开启、结束的日期和时间的电子数据
[3]每一生产工序的负责人的电子签名;
[4]生产步骤操作人员的电子签名;必要时,还应当有操作(如称量)复核人员的电子签名;
[5]每一原辅料的批号以及实际称量的电子数据,包括物料消耗。
[6]从生产设备、控制系统上采集的关键工艺参数和人工操作记录(如人工观察、清场、检查)。记录主要生产设备的编号、名称。
[7]中间控制结果的电子记录以及操作人员的电子签名;
[8]不同生产工序所得产量的电子数据及必要时的物料平衡自动计算;
[9]对特殊问题或异常事件的电子记录,包括对偏离工艺规程的偏差情况的详细说明,并经授权人员的电子签字批准。
1.1.1包装操作
电子批记录的包装部分应当有待包装产品的批号、数量以及成品的批号和计划数量。
应当使用信息化手段记录包装开始前的检查,确保工作场所、包装生产线及其他设备已处于清洁或待用状态,无上批遗留的产品、文件或与本批产品包装无关的物料。检查结果应形成电子记录。
应当基于批准的包装材料版本,核对包装材料正确无误,核对待包装产品和所用包装材料的名称、规格、数量、质量状态。核对结果应形成电子记录。
电子批包装记录的应当显示所包装产品的名称、规格、包装形式和批号。
电子批包装记录至少要包含以下内容:
[1]产品名称、规格、包装形式、批号、生产日期和有效期的电子数据
[2]包装操作的日期和时间的电子数据
[3]包装操作负责人的电子签名;
[4]包装工序的操作人员的电子签名;
[5]每一包装材料的名称、批号和实际使用数据的电子数据,包括包装材料的消耗。
[6]从包装设备、控制系统上采集的关键工艺参数和人工操作记录(如人工观察、清场、检查)。记录主要包装设备的编号、名称。
[7]检查记录、中间控制结果的电子记录以及检查人员的电子签名;
[8]所有印刷包装材料的实样与电子记录副本,并印有批号、有效期及其他打印内容;不易随批包装记录归档的印刷包装材料可采用印有上述内容的图像或扫描件。
[9]对特殊问题或异常事件的电子记录,包括对偏离工艺规程的偏差情况的详细说明或调查报告,并经授权人员的电子签字批准。
[10]所有印刷包装材料和待包装产品的名称、代码,以及发放、使用、销毁或退库的数量、实际产量以及物料平衡检查的电子记录;
1.2检验记录
应当采用电子检验记录包含《药品生产质量管理规范》中对检验记录的所有内容。
电子化检验记录应当包括中间产品、待包装产品和成品的质量检验记录,并可追溯该批疫苗所有相关的质量检验情况。
应当通过自动采集并记录检验过程中产生的相关数据,同时允许分配了操作权限的人员把人工操作、观察的信息录入系统;
电子检验记录的检验类型和范围,应按预先设置的公式进行计算和修约,如采用计算机化系统的自动复核,该功能应经过验证。
已生成的电子检验记录应可进行查阅和导出,并确保相关数据的真实、完整和可追溯。
疫苗上市许可持有人可根据疫苗生产工艺的特性以及实际生产、检验情况,基于质量风险管理的原则,充分识别生产与检验过程中的关键工艺参数和关键质量属性,形成关键数据项并采用信息化手段进行监控。以下疫苗关键数据项示例,供参考。
建议疫苗上市许可持有人按照《疫苗追溯基本数据集》(NMPAB/T1004-2019)中对数据集内容的要求,对关键数据项进行整理,逐步积累和建设完整的适用监管的数据集。


甚至,我们在很早官方颁布的《国家药品监督管理局信息化标准》NMPAB/T1004—2019中就已经看到所有剂型都有对应标准电子化编码。
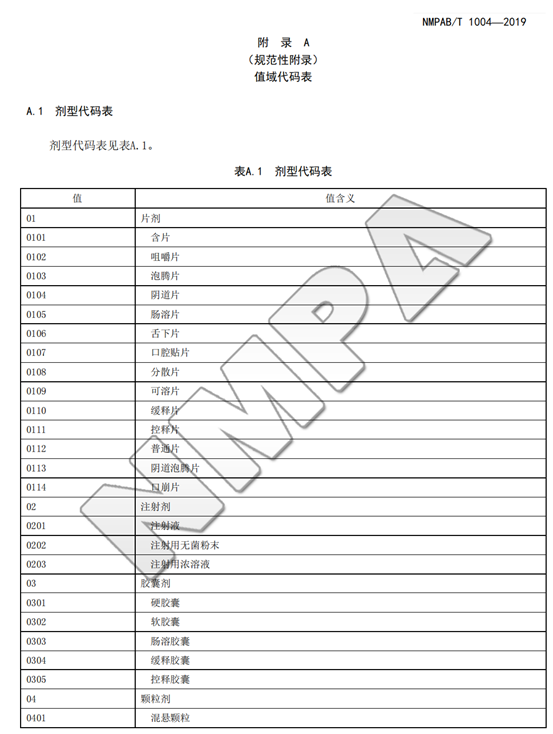
与其说对于欧盟某个GMP附录修订和生效非常关注,不如重新梳理和阅读我们国内2019-2022颁布的官方技术规范和指南。
崇洋媚外的说法可能有些过分,但是一叶障目不见泰山的现状需要改改了。
关于举办中药饮片炮制技术研究与质量控
各会员单位: 为顺应药品监管规..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


