

在过去的一个世纪里,治疗药物已经从植物提取物发展到了化学定义的小分子,又发展到了基于大分子的重组蛋白。经过几十年不懈的科学努力,前景被设想了几十年的基因和细胞治疗,现在已开始推动治疗药物格局的下一步改变,为此前无法解决的疾病开辟了全新的治疗途径。目前获批的多种此类治疗产品包括用于细胞治疗的Maci、Kymriah、Yescarta和Tecartus,以及用于体内基因治疗的Luxturna和Zolgensma®。这一系列的疗法一起促成了数百项临床试验的启动,这些试验预示着在未来十年内可能将出现更为成功的治疗方法。Luxturna治疗视网膜营养不良,Zolgensma®治疗脊髓性肌肉萎缩,其都是具有突破性的治疗方法,患者数量虽少,但非常重要。随着基因治疗的成熟,更常见的疾病,如实体瘤,将以更大的患者群体出现,作为此类产品的治疗对象。
为了更好地利用基因和细胞治疗对于常见疾病的明确且巨大潜力,其制造过程需要提高生产力和效率。早期的制造工艺是在科研实验室中开发的,使用的技术不是为工业性规模放大而设计的。在这份专门针对基因治疗的白皮书中,我们将这种用于早期基因治疗临床和商业化批次的前期工作称为基因治疗制造1.0 (GTM 1.0)。早期基因治疗的过早成功,加上GTM 1.0的局限性,已经造成了一个巨大的供应链缺口,病毒载体的需求超过了当前的供应量,而且这个缺口还在扩大。行业迫切需要可提高生产力和效率的重大技术发展,以满足当前的需求,并推动未来的工作。本文总结了一些GTM 1.0面临的挑战,并将介绍潜在的基因治疗制造2.0(GTM 2.0)在生产力、可放大性和安全性方面的改进。
制造需求缺口
决定基因治疗需求的大量变量,包括适应症、处方剂量和患者数量,阻碍了明确未来价值的计算。不过,为了更好地理解不同的复杂情况,一些研究人员已经建立了预测模型。来自麻省理工学院(MIT)的Quinn等人开发了一个基线模型,用于预测针对不同疾病的获批疗法的数量和患者群体(1)。根据条件的差异,每种疗法的患者人数可从100人(如眼科治疗)到超过20,000人(如神经治疗)不等。该模型的输出估计“到2030年,可能会有35万患者使用30到60种不同的基因疗法进行治疗。”据估计,到2030年,每年可治疗的患者数量将达到5万人,很可能至少是目前患者数量的10倍。在第二个模型中,Rininger等人(2)通过编排一系列治疗适应症中每个患者的剂量,估算了AAV和慢病毒载体的生产需求和产能,从而可以推算出AAV和慢病毒生产的年需求量。例如,他们估计血液病适应症的平均剂量为每位患者5 x 10^8 TU慢病毒。假设我们考虑每年至少治疗15,000名患有这种疾病的患者,那么每年就需要生产7.5 x 10^12 TU的慢病毒。对于肌萎缩症,作者估计平均剂量为每人3.9 × 10^15 VG AAV。在美国,5 - 24岁的男性约有1/5600-7700的发病率(3),世界范围内约有1万名杜氏肌萎缩症患者,这将需要大约4 × 10^19 VG AAV。尽管Quinn等人的模型和Rininger等人的模式使用不同的目标、策略和假设构建,但得出相同的结论;病毒载体的供需缺口要么已经存在,要么很快就会出现。
病毒载体供需缺口源于这样一个事实:病毒载体制造技术和工艺主要是由重组蛋白和单克隆抗体(mAb)工艺重新利用而来,而非优化。将投放市场的时间优先于技术和工艺优化,加剧了供需之间的差距。耗时的优化迭代过程在很大程度上被放弃了,因为只要生产出所需数量的病毒载体,低产量工业通常也会被认为是“足够好了”。而在之后的商业化制造中,这就成为了一个重要的工艺瓶颈。
短缺的影响因指标的不同而有很大差异。例如,一种血液肿瘤的总制造需求,估计为每年需要7.5 x 10^12 TU慢病毒,可能可以通过现有的GTM 1.0技术来满足。一个总收率为30%的100 m2固定床贴壁生物反应器,一年内运行20次,很可能满足这样的需求。
但考虑到每年会有1万名肌肉萎缩症患者时,情况就不一样了。同样,使用估计的数字,一个3,000 L的STR每年可进行20批,产量为1 × 10^10 AAV vg/day/mL,总工艺收率为30%,即可提供~5 × 10^17 VG AAV。为了满足4 × 10^19的要求,需要80个3,000 L生物反应器,每个反应器每年运行20个批次,这是不合理的。
病毒载体产量的每一个数量级的增加都将为该领域创造新的机会。产量增加10倍将显著降低成本,扩大现有疗法的可及性,使患者能够更公平地获得治疗,并扩大全球渠道。100倍的提升将把适应症,如肌肉萎缩和其它疾病,带入到可实现的制造方案范畴内。最后,1,000倍的提升可以让基因治疗进入一个新时代,在这个时代里,它可以治疗患者群体数量更大的常见疾病,并彻底改变现代医疗模式。
瞬时表达和贴壁细胞培养受限的生产力和可放大性
目前的上游病毒载体制造过程主要基于瞬时表达技术,虽然其在实验室环境中较为方便进行,但通常存在产率低且操作复杂等问题,可能会对制造的可重复性造成挑战。对于AAV和慢病毒,利用瞬时表达,分别有报道获得了1 - 5 × 10^4 VG/cell(5)和1 - 10 TU/cell (6)的细胞特异性产率。大多数系统需要3到4个DNA质粒,每个都具有特定的复杂性,需要对多个质粒进行多个参数的优化,如DNA与转染试剂的比例、DNA与细胞的比例以及孵育时间,这会增加执行DOE所需的资源量,也会增加单元操作执行过程中发生偏差的风险。此外,转染试剂会影响细胞生长,转染后活细胞密度一般会下降,这意味着能够实现转染的试剂也会降低可用于生产的细胞。这种复杂性在为临床应用制造高质量质粒时,将转化为巨大的成本,导致多质粒转染成为整体工艺成本的重要因素(4)。
通过提高对细胞代谢和蛋白质分泌的理解,重组蛋白和单克隆抗体的细胞特异性产率已经实现了数倍的提升。将这种方法扩展到基因治疗将是针对供需缺口的有效解决方案,但其不太可能赶上目前临床管线的快速时间线,因为,目前单克隆抗体细胞特异性产率10 - 100倍的提升花了大约20年的时间。鉴于病毒载体在生物化学上比大多数重组蛋白和单克隆抗体更为复杂,要使病毒载体细胞特异性产率获得相当的提升,预计至少需要相似的时间。此外,考虑到代谢和病毒载体分子的细微差别,针对某一产品的细胞特异性产率解决方案不太可能广泛适用。
贴壁细胞培养工艺是一种在实验室规模条件下获得基因治疗产品的有效的方法,而其在生产规模条件下,最终将遇到不小的挑战。虽然贴壁培养很容易生长,且可通过培养基置换而维持,但其细胞密度通常较低(~1 × 10^6 cells/mL),且受限于扁平培养容器的尺寸。市面上最大的平面培养设备(~2.5 m2, ~6 L培养基体积)对于AAV和慢病毒,分别可提供不高于3 × 10^14VG和6 × 10^10 TU的产量。将设备面积增加100 X,至250 m2或以上,以满足需求差距,并不是一种实用或可执行的解决方案。与规模放大不同的是,使用大量小型容器进行规模扩展有其自身的障碍,包括:自动化所需的设备成本高昂、复杂性增加、生产占地以及劳动力需求。作为向前发展的一步,新一代固定床生物反应器已被证明可以将每个生物反应器的病毒载体产量提高100X,如对于AAV,可达~2 × 10^16 VG(7),对于慢病毒,可达~1 × 10^12 TU(8),但这仍然不足以满足需求。目前最大的固定床生物反应器只到500 m2,放大到相当于2,000 – 3,000 L STR的规模仍然无法实现。
非优化的下游导致低收率
从生物药工艺到病毒载体工艺的过滤和层析方法的转化确实可实现所需产品的生产,但回收率远低于预期水平。单抗的整体下游收率通常能达到80%,而病毒载体通常只能达到30%,有些甚至低至10%。与蛋白质相比,病毒载体具有更大的结构和生化复杂性,可应用的方法更少,稳定的缓冲液和温度条件选择更有限。例如,通常用于蛋白质的低pH值和高电导缓冲条件会导致病毒失活,如脆弱的、有囊膜的慢病毒。大多数产品损失发生在非优化的层析分离方法,需要在纯度和收率之间进行折衷。
亲和填料能有效地去除AAV中的宿主细胞蛋白(HCP)和DNA杂质,但受到AAV血清型多样性的挑战,并不是所有的衣壳都能与目前市售的AAV填料结合,而能够结合的衣壳的回收率也各不相同,公布的结果在50% - 90%之间(9、10)。
AAV精制步骤主要作用是将缺乏DNA(空)的衣壳从含有DNA(完整)的衣壳中分离出来。核酸被包裹在衣壳内,它的存在可改变整个病毒的有效等电点,在大多数情况下,至少可以通过离子交换部分分离两种物质。然而,部分分离要求在产量和纯度之间进行操作选择;在完整衣壳纯度较低的情况下,可能获得较高的总收率;反之亦然,更高纯度的完整衣壳一般伴随着较低的总收率。由于纯度通常被优先考虑,这一关键步骤的收率会受到影响。使用离子交换层析从完整衣壳中分离空衣壳也增加了开发、验证和规模放大的时间,原因在于多方面的变量,包含但不限于,形式(填料、膜和整体柱)、化学(不同配基密度的强或弱阴离子和阳离子基团)、以及分离条件(pH、电导率、缓冲液组成、梯度或步骤洗脱)。
目前还没有开发用于慢病毒的亲和配基,使离子交换层析成为了主要的方法,而体积排阻层析(SEC)是次优的第二方法。在收获和初步处理后,通过基于膜的离子交换吸附剂进行澄清,抛弃传统的UF/DF浓缩和洗滤,为慢病毒纯化提供了另一种解决方案。膜层析后较小的洗脱体积允许第二步的柱层析以较慢的流速进行。下游工艺中的过滤步骤,如使用深层过滤(11,12)进行的澄清和使用切向流过滤(TFF)(13)进行的产物浓缩和制剂,也经常会导致产物损失。吸附至过滤介质、剪切应力和滞留体积都是工艺过程中产物损失或失活的原因。
工艺安全性
GTM 1.0技术缺乏污染物去除和确保工艺安全性的适当工具。由于产品本身是病毒或病毒载体,治疗性蛋白质污染物(细菌或外来病毒)去除技术,如低pH值病毒灭活和除菌/除病毒过滤,并不总是兼容的。这就迫切需要防止污染而不是去除污染。
此外,用于在线监测产品关键质量属性(CQA)的分析技术在基因治疗中也有限。一般来说,随着分子复杂性的增加,分析方法也将变得更加复杂。从小分子到重组蛋白,质谱等方法用了几十年的时间才适应新的要求。基因和细胞治疗需要对现有方法和新的分析技术进行类似的调整。由于pH值、电导率和压力条件不能直接检测基因和细胞治疗产品的质量,因此将需要新的方法在线监测关键质量属性(CQA)。大多数基因治疗分析方法都是离线进行的,其中一些方法,如通过透射电子显微镜(TEM)定量检测AAV衣壳,可能需要长达两周的时间。
向基因治疗制造2.0跃进
生产细胞系强化悬浮细胞培养的优势
多家基因治疗公司已经明确有必要升级GTM 1.0,以填补供需缺口,这些公司正在迅速采用和实施创新技术,以快速过渡到GTM 2.0,并已取得一些重大成果。
GTM 2.0的大部分工作集中在提高细胞培养效率,使用的方法与开发用于治疗性蛋白质的方法相似。许多过往的改进,如培养基优化和补液策略,已获得了显著的增量收益,但不是革命性的改变。基因治疗需要实现的最关键的细胞培养步骤变化,同样也发生在重组蛋白的生产中,即将贴壁细胞转变为悬浮模式。虽然贴壁细胞提供了快速获取适量物料的简易收获方法,并可降低杂质,但规模放大的挑战妨碍了其在临床或商业化生产阶段的广泛适用性。Repligen的数据显示,60%的基因治疗公司已经从贴壁平台过渡到了悬浮模式,30%的公司同时支持两个平台,10%的公司仍然坚持贴壁模式。对悬浮细胞的投资可以为促进工艺规模放大、扩大细胞培养体积以及提高病毒滴度提供显著的好处。悬浮细胞也增加了一个安全的因素,因为它们通常生长在无血清的化学限定培养基中。然而,在悬浮模式中,观察到的病毒滴度通常较低,因此需要使用其它方案来进一步优化工艺。
细胞截留装置,如XCell ATF®技术,可以提高生物反应器活细胞密度(VCD)至300 × 10^6 cells/ml以上,而这可通过几种不同的方式,用于提高工厂的整体产能。当连接到生产生物反应器时,细胞截留装置可以应用于以离散或连续模式提高产量。在离散模式下,细胞截留装置能够在确定的时间点从单个生物反应器中进行多次收获,通常称为“补料分批强化”。在连续灌流模式下,产物在整个生产过程中从生物反应器中获得,通常称为“灌流”。在任何一种模式中,生产生物反应器的产量将增加,且停机时间降低。
细胞截留装置通过同时执行收获和澄清步骤,可进一步强化上游操作。每个细胞截留装置包含一个孔径0.2 - 0.65 μm的过滤器,条件培养基通过过滤器进入滤液。因此,收获的物料可以直接从收获进入层析操作,而不需要离心或深层过滤,形成了额外的工艺效率。
细胞截留装置也可用于提高种子扩增生物反应器的细胞密度。美国Cytovance® Biologics最近的一项研究表明,通过提高VCD,在病毒载体生产工艺中,产量提高了一个数量级。标准细胞培养的VCD可达到~1.3 × 10^6 cells/ml,而强化工艺的VCD提高了10倍,达到~1.2 × 10^7 cells/ml。VCD的提高意味着收获的衣壳量的提高。标准进料分批工艺可产生1-3 × 10^10 capsids/ml,而XCell ATF®强化工艺的产量提高了大约100倍,达到4-10 × 10^12 capsids/ml。
图1.使用XCell ATF®进行的AAV2生产工艺的强化(数据来自(14),由Cytovance® Biologics提供)。使用XCell ATF®细胞强化装置进行的灌流工艺(转染前)的AAV2产量相比标准批次工艺提高了>100倍的衣壳生产。
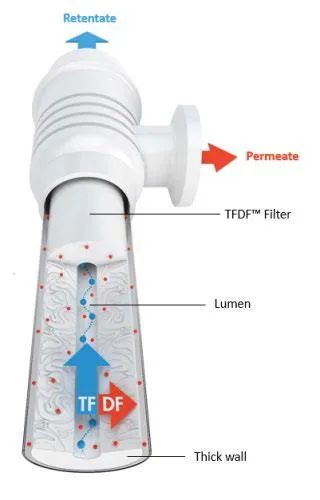
图2.切向流深层过滤(TFDF®)以切向模式运行深层过滤器。细胞培养基通过管状深层过滤器的管腔。滤液通过深层过滤器管壁,并从滤液端口流出。回流液离开管腔并直接返回至生物反应器。
使用新型TFDF®技术也可以实现细胞截留,该技术已经证明了其在提高慢病毒产率方面的卓越性能。TFDF®技术将切向流和深层过滤结合在一次性使用且封闭的过滤装置内。具有2-5 μm孔径的厚壁管状深层过滤器能够捕获细胞和细胞碎片,产物截留可忽略不计。切向流通过引导大部分细胞和细胞碎片朝向回流端流动并远离过滤器,从而降低污染(图2)。切向流和深层过滤的空前结合,可获得高度澄清的溶液,且可稳健对抗污染。Oxford Biomedica最近发表的一篇文章报道,与标准批次收获相比,使用TFDF®,慢病毒产量提高了两倍以上(15)。对于标准补料分批生物反应器的单次收获,深层过滤收率为~70%,而TFDF®的收率为~90%。更重要的是,由于TFDF®在低剪应力的切向模式下操作,而不是作为一个死端过滤器,完整的细胞被截留在生物反应器中。用新鲜培养基补充生物反应器,可使细胞能够继续产生病毒,并在几天后进行第二次收获。与基于深层过滤的单次收获工艺相比,每次收获更高的收率与二次收获的综合效益,使TFDF®工艺的产量达到了原始工艺的~250%。TFDF®技术还可以降低病毒失活的风险,其通过一系列的快速收获,从生物反应器中取出产物,而不是在高细胞密度、高温度的生物反应器中积累慢病毒。
到目前为止,细胞培养强化主要应用于瞬时系统中转染前的细胞扩增。稳定细胞系的开发,将消除质粒转染的需求,有助于实现细胞培养强化的全部优势。已经有文章报道过慢病毒(6、16)和AAV(9)稳定细胞系的早期案例。含病毒衣壳基因的包装细胞系也被认为是减少对转染步骤依赖性的一种可行方法,虽然其仍需要转染携带目的基因的质粒,但与目前的多质粒转染工艺相比,单质粒转染将大大简化工艺,降低整体生产时间。从转染贴壁细胞瞬时表达到稳定表达悬浮平台的过渡可实现GTM 2.0定义所需的10至100倍的上游产量提升。
上游病毒载体产量的数量级提升将需要下游工艺中相应的步骤变化改进。澄清,一个经常被忽视的工艺强化机会,现在可以通过多种技术来提高产量,减少资源需求,或直接将收获与澄清相结合。TFDF®是最新开发的过滤模式之一,其可用于澄清步骤(以及生物反应器强化)。通过引导绝大部分细胞通过循环回路返回至生物反应器,而不是进入死端深层过滤器,TFDF®极大地提高了过滤能力和通量(图3)。增加的过滤能力允许TFDF®技术使用较低的过滤表面积澄清高密度细胞培养液,随着细胞密度的不断增加,这一点非常重要。简单地放大传统深层过滤器的表面积,以适应更高的生物反应器细胞密度,将显著提高澄清资源要求,包括操作时间、成本、缓冲液、WFI、车间空间以及GMP存储空间。将过滤器的尺寸缩小10倍,可以在不同的具体情况下,缓解上述每一个考虑因素,不仅可以在某一工艺的强化中扮演重要的角色,还可以强化整个工厂,以使每天、每平方英尺工厂空间可提供更多的产品。此外,TFDF®过滤器采用伽马辐照的整合式干型流路形式发货,不需要使用WFI或缓冲液冲洗。

图3. KrosFlo® TFDF®系统使用循环回路,使细胞培养液和细胞从生物反应器流出,通过TFDF®过滤器的管腔,再返回到生物反应器。进入管状深层过滤器壁的细胞数量降低,从而显著提高了过滤能力。
用于快速下游工艺开发的高通量工具
针对基因治疗产品的下游技术的开发将是非常有益的,但由于它们不太可能在未来两到三年内实现,因此需要快速优化现有的方法来实现这种新应用。高通量工具提供了一种可用且易于实施的方法,以弥补由于对进入市场的速度进行优先级排序而导致分配给下游工艺优化的时间不足的问题。微型预装层析柱,OPUS® RoboColumn®层析柱是应用最广泛的一种方式,其可以使用96孔板形式和机器人液体处理工作站实现自动化层析工艺的开发。大量不同的填料、化学材质和纯化工艺参数可以在几个小时内完成筛选,且每个实验仅需要微升级样品体积。预装层析柱在工艺放大过程中继续扮演着关键角色。具有高批次可再现性的高质量层析柱可节省操作人员的准备时间,但更重要的是,便于验证,提高日程安排的灵活性,加速技术转移,最终为整个项目节省大量时间和资本支出。
用于产物浓缩和制剂的TFF的优化从选择中空纤维还是平板膜包过滤器形式开始。产物对剪切的敏感性是影响这一决策的关键参数。中空纤维由于其开放式结构,产生的剪切更低。中空纤维形式通常是囊膜病毒工艺的首选,如慢病毒,因其很容易因剪切过高而失活。对于更为稳定的病毒,如AAV,膜包形式同样有效,且可能可缩短工艺时间,甚至可以在有限的过滤表面积条件下提高滤液通量。确定膜或纤维的截留分子量(MWCO)决定了筛分效果,继而决定了收率以及杂质去除水平。MWCO 500 ~ 750 kDa 的膜可有效地处理慢病毒(~120 nm)等大尺寸的病毒载体。而较小的病毒,如AAV (~25 nm)与MWCO在100 ~ 300 kDa之间的膜配合最好。然后,工艺参数(跨膜压或TMP、错流流速、通量)的优化可以最大限度地降低工艺时间和所需的膜表面积。与适当规模的自动化系统硬件选择相结合,可通过减少人为错误,而使收率最大化,降低时间需求和工艺偏差。
用于提高工艺安全性的封闭式、一次性使用流路
维持单元操作尽可能封闭有助于降低污染的风险。伽玛辐照、一次性使用、预组装的工艺流路提供了一种低生物负荷、方便且可立即实施的污染缓解解决方案。供应链生产多样化配置的能力正在扩大。例如,ProConnex® 流路可以从超过250个部件的部件库选择部件进行构建,包括管路、储袋和/或容器、过滤器、传感器、泵头和无菌接头。流路设计的灵活性使封闭式系统的优点能够应对于整个工作流中广泛的工艺变化。
用于优化工艺监测的更好的分析技术
用于在线工艺监测的过程分析技术可帮助实现工艺的安全性、操作的方便性,也使工艺更加稳健。由于化学组成的性质,病毒载体具有不同于单抗和蛋白质的CQA,因此需要不同的CQA监测分析技术。治疗性蛋白工艺中常用的工艺参数,如压力、pH值、电导率和UV也可用于病毒载体工艺,但它们不是产品稳定性或纯度的有效指标。
例如,UF/DF单元操作过程中的蛋白质浓度通常是通过从流路中提取样品,然后使用比色皿进行耗时且容易出错的UV-Vis分光光度计检测来确定的。这种方法在浓缩单元操作结束时会产生不小的挑战,因为体积的小变化可能会导致产物浓度的大变化。取样、稀释样品和检测浓度的繁琐、步进式过程迫使操作者必须在工艺控制和工艺时间之间进行权衡。如果工艺控制为优先级,那么将停止浓缩,直到获得可用的分析数据,这会导致整个完成时间的延长。或者,如果工艺时间为优先级,那么在分析过程中将允许浓缩继续,这会在实验流路中的实际产物浓度与UV-Vis分光光度计中分析的产物浓度之间产生一个滞后,导致过度浓缩。
Slope Spectroscopy®(斜率光谱法)技术的开发大大简化了UV-Vis光谱法检测,其消除了样品制备过程中对连续稀释的需求。Beer-Lambert公式(A=e*C*l)将吸光度(a)描述为消光系数(e)、浓度(c)和光程(l)的乘积。Slope Spectroscopy®技术改变光程而保持样品浓度固定,而传统的UV-Vis方法维持光程固定,而改变样品浓度(通过连续稀释)。简化分析方法的这一部分将在方法和程序级别上形成多个级联优势。UV-Vis标准操作程序(SOP)文件长度显著减少,获得结果的时间可从1-2小时缩短至5-10分钟。更快的结果获得时间几乎可允许立即访问分析数据,以做出关键决策,使浓缩单元操作在更稳健的监测条件下,快速完成。对于需在多个组和/或地点之间转移的工艺,这一精简且稳健的单步骤操作可通过消除可能存在差异的操作人员移液和连续稀释步骤,而节省数月的技术转移时间。
最近,Slope Spectroscopy®技术在向GTM2.0发展的道路上又迈出了重要一步,其经工程设计,而可作为在线仪器,与UF/DF流路相整合(图4)。在线监测可以实现无与伦比的工艺控制,这是影响病毒载体等治疗药物稳定性的一个重要因素。使用FlowVPE® Slope Spectrometer®在AAV浓缩和洗滤单元操作中获得的初步数据表明,在线获得的UV-Vis数据与离线获得的数字液滴QPCR(ddQPCR)之间存在良好的相关性(图5)。
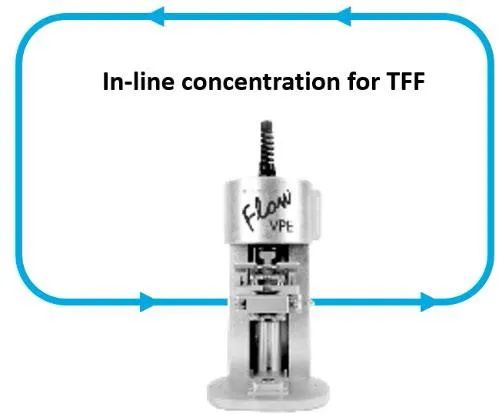
图4. Flow VPE®设备整合至TFF流路,用于在线监测产物浓度。
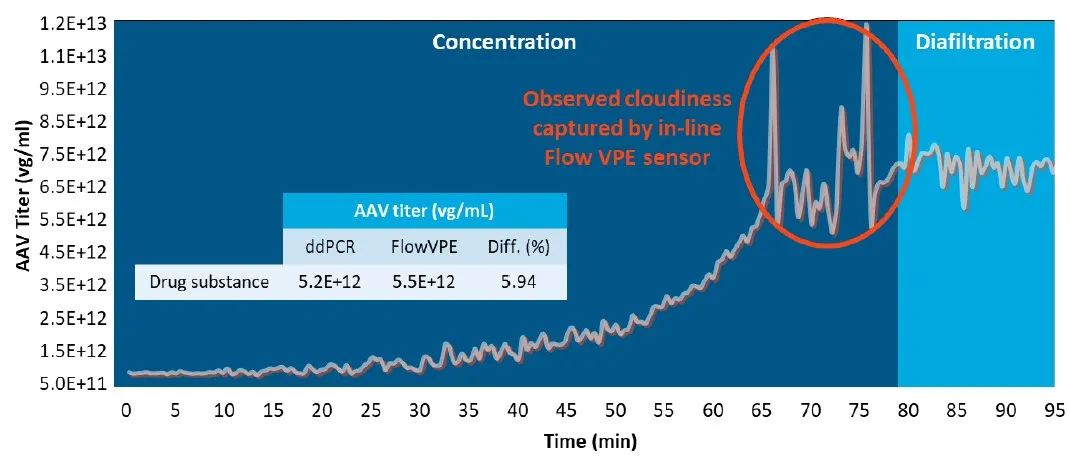
图5. 使用Slope Spectroscopy® FlowVPE®技术,在最终浓缩和洗滤工艺步骤中,进行AAV滴度的在线监测。在线监测可以在发生工艺偏差时进行快速调整(例如,观察到浑浊现象,其表明可能存在病毒聚集,需要停止浓缩和/或改变缓冲液)。
从0到65分钟,对浓缩步骤进行监测,滴度从1.0 x 10^12 vg/ml稳定增加到大约6.0 x 10^12 vg/ml。平滑的增长曲线在65分钟时出现偏离,形成了一个信号尖峰,突出了在线监测的价值,其与表明出现病毒聚集的样品浑浊一致。结合此观察结果,在80分钟时开始洗滤。在早期开发中,针对SOP,准确确定临界聚集或沉淀浓度可以节省大量的时间和物料。在生产环境中,实时数据的获取可以提高产品质量,降低工艺偏差,并加快整个工艺时间。
总结
使第一批具有里程碑意义的基因疗法成功完成临床试验和监管审批的生产工艺缺乏满足当前需求所需的效率和产量,更不用说未来的需求了。这些工艺,这里称为基因治疗制造1.0 (GTM 1.0),受限于瞬时表达、贴壁细胞培养、低纯化收率以及离线检测CQA的生产力和可放大性挑战。现有方法的工艺优化和新技术的创新将引领我们走向基因治疗制造2.0,整体生产力将提高10到100倍。稳定细胞系产生细胞,灌流强化的悬浮细胞培养、产物特异性亲和填料、完全封闭的下游操作以及新的分析技术将很可能被纳入其中。未来五年内,基因治疗制造2.0的出现将有助于确保基因治疗供应链的稳定性,并帮助解决目前尚未解决的、危及生命的疾病。
参考文献:
Quinn C. et al., Estimating the clinical pipelineof cell and gene therapies and their potential economic impact on the US healthcare system, Value Health, 2019; 22(6):621–626.
Rininger J. et al., Capacity analysis for viral vector manufacturing: Is there enough?, BioProcess Int., Nov-Dec 2019,17(11–12)s.
Centers for Disease Control and Prevention.(2009). Prevalence of Duchenne/Becker muscular dystrophy among males aged 5-24years—four states, 2007. MMWR Morbidity and Mortality Weekly Report, 58,1119 - 1122. Retrieved June 21, 2012, fromhttp://www.cdc.gov/mmwr/preview/mmwrhtml/mm5840a1.htm.
Cameau E., Pedregal A. and Glover C., Cost modelling comparison of adherent multi-trays with suspension and fixed-bed bioreactors for the manufacturing of gene therapy products, Cell & Gene Therapy Insights 2019; 5(11), 1663–1675.
Grieger J., Soltys S. and Samulski R., Productionof recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1clinical vector, Molecular Therapy 2016, 24(2), 287–297.
Manceur A. et al., Scalable Lentiviral Vector Production Using Stable HEK293SF, Human Gene Therapy Methods 2017, 28(6),330-339.
Lefebvre P. et al., Development of a Scalable Viral Vector Upstream Process for Gene Therapy: rAAV-8 Production by Transient Transfection of HEK-293 Cells in iCELLis® Bioreactor, Poster Presented at ESACT2017.
Valkama AJ et al., Optimization of lentiviral vector production for scale-up in fixed-bed bioreactor, Gene Therapy 2018, 25,39–46.
Nass S. et al., Universal method for the purification of recombinant AAV vectors of differing serotypes, Mol Ther Methods Clin Dev. 2017 Dec 22;9:33-46.
Toueille M. et al., Development of purification steps for several AAV serotypes using POROS™ CaptureSelect™ AAVX, Gene Therapy Insights 2018; 4(7), 637-645.
Moss D., Vector purification: issues and challenges with currently available technologies, Cell Gene Ther. Ins. 2019; 5(9):1125–32.
Raghavan B. et al., Optimizing the clarification ofindustrial scale viral vector culture for gene therapy. Cell Gene Ther. Ins.2019; 5(9): 1311–22.
Ruscic J. et al., Lentiviral vector purification using nanofiber ion-exchange chromatography, Molecular Therapy: Methods & Clinical Development 2019, 15, 52-62.
Short C., High Cell Density Perfusion for the Production ofAAV2 Viral Vector Using the Repligen XCell ATF System, Repligen Technical Seminar, Burlingame, CA, Oct 17th,2019.
Williams T. et al., Lentiviral vector manufacturing process enhancement utilizing TFDF™ technology, Cell & Gene Therapy Insights 2020; 6(3), 455–467.
Carrondo M. et al., LentiPro stable producer cells:Delivering scalable and reliable lentiviral vector manufacturing, in "Advancing Manufacture of Cell and Gene Therapies VI", Dolores Baksh, GE Healthcare, USA Rod Rietze, Novartis, USA Ivan Wall, Aston University, United Kingdom Eds, ECI Symposium Series, (2019).
关于举办中药饮片炮制技术研究与质量控
各会员单位: 为顺应药品监管规..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


