

根据《保健食品注册管理办法(试行)》,为规范、统一营养素补充剂等申报与审评行为,我局制定了《营养素补充剂申报与审评规定(试行)》、《真菌类保健食品申报与审评规定(试行)》、《益生菌类保健食品申报与审评规定(试行)》、《核酸类保健食品申报与审评规定(试行)》、《野生动植物类保健食品申报与审评规定(试行)》、《氨基酸螯合物等保健食品申报与审评规定(试行)》、《应用大孔吸附树脂分离纯化工艺生产的保健食品申报与审评规定(试行)》、《保健食品申报与审评补充规定(试行)》8个与保健食品申报与审批相关的规定。上述规定于2005年7月1日起正式实施,现予以通告。
国家食品药品监督管理局
二○○五年五月二十日
营养素补充剂申报与审评规定(试行)
第一条 为规范营养素补充剂的审评工作,根据《中华人民共和国食品卫生法》和《保健食品注册管理办法(试行)》,制定本规定。
第二条 营养素补充剂是指以补充维生素、矿物质而不以提供能量为目的的产品。其作用是补充膳食供给的不足,预防营养缺乏和降低发生某些慢性退行性疾病的危险性。
第三条 营养素补充剂必须符合下列要求:
(一)仅限于补充维生素和矿物质。维生素和矿物质的种类应当符合《维生素、矿物质种类和用量》的规定。
(二)《维生素、矿物质化合物名单》中的物品可作为营养素补充剂的原料来源;从食物的可食部分提取的维生素和矿物质,不得含有达到作用剂量的其他生物活性物质。
(三)辅料应当仅以满足产品工艺需要或改善产品色、香、味为目的,并且应当符合相应的国家标准。
(四)适宜人群为成人的,其维生素、矿物质的每日推荐摄入量应当符合《维生素、矿物质种类和用量》的规定;适宜人群为孕妇、乳母以及18岁以下人群的,其维生素、矿物质每日推荐摄入量应控制在我国该人群该种营养素推荐摄入量(RNIs或AIs)的1/3~2/3水平。
(五)产品每日推荐摄入的总量应当较小,其主要形式为片剂、胶囊、颗粒剂或口服液。颗粒剂每日食用量不得超过20克,口服液每日食用量不得超过30毫升。
第四条 使用《维生素、矿物质化合物名单》以内的物品,其生产原料、工艺和质量标准符合国家有关规定的,一般不要求提供安全性毒理学试验报告;使用《维生素、矿物质化合物名单》以外的物品,应当提供该原料的营养学作用、在人体内代谢过程和人体安全摄入量等科学文献资料以及依照新资源食品安全评价的有关要求出具的安全性毒理学评价试验报告。
第五条 申请人应当提供营养素补充剂中营养素的定量检验方法。
第六条 营养素补充剂标示值是指产品标签和说明书上所标示的该产品中某种营养素含量的确定数值,不得标示为范围值。
营养素补充剂标示值以及产品质量标准中营养素含量范围值应符合本规定第三条第四款的有关规定。
第七条 含有三种以上维生素或矿物质的营养素补充剂,方可称为多种维生素或矿物质补充剂。
第八条 产品应采用定型包装,便于消费者食用和保持产品的稳定性,直接与营养素补充剂接触的包装材料必须符合有关卫生标准或卫生要求的规定。
第九条 营养素补充剂标签、说明书应符合国家有关规定,同时还应当标明以下内容:
(一)“营养素补充剂”字样。
(二)营养成分应当标示最小食用单元的营养素含量。
(三)食用方法及食用量,应当明确不同人群具体推荐摄入量。
(四)注意事项,应当明确产品不能代替药物,不宜超过推荐量或与同类营养素补充剂同时食用。
第十条 《维生素、矿物质的种类和用量》、《维生素、矿物质化合物名单》由国家食品药品监督管理局制定并发布。
第十一条 本规定由国家食品药品监督管理局负责解释。
第十二条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
表1:维生素、矿物质种类和用量
表2:维生素、矿物质化合物名单


真菌类保健食品申报与审评规定(试行)
第一条 为规范真菌类保健食品审评工作,确保真菌类保健食品的食用安全,根据《中华人民共和国食品卫生法》、《保健食品注册管理办法(试行)》,制定本规定。
第二条 真菌类保健食品系指利用可食大型真菌和小型丝状真菌的子实体或菌丝体生产的产品。真菌类保健食品必须安全可靠,即食用安全,无毒无害,生产用菌种的生物学、遗传学、功效学特性明确和稳定。
第三条 除长期袭用的可食真菌的子实体及其菌丝体外,可用于保健食品的真菌菌种名单由国家食品药品监督管理局公布。
第四条 国家食品药品监督管理局对保健食品的真菌菌种鉴定单位进行确定,确定的菌种鉴定单位的名单由国家食品药品监督管理局公布。
真菌类保健食品的菌种鉴定工作应在国家食品药品监督管理局确定的鉴定单位进行。
第五条 申请真菌类保健食品,除按保健食品注册管理的有关规定提交资料外,还应提供以下资料:
(一)产品配方及配方依据中应包括确定的菌种属名、种名及菌株号。菌种的属名、种名应有对应的拉丁学名。
(二)菌种的培养条件(培养基、培养温度等)。
(三)菌种来源及国内外安全食用资料。
(四)国家食品药品监督管理局确定的鉴定机构出具的菌种鉴定报告。
(五)菌种的安全性评价资料(包括毒力试验)。菌种及其代谢产物必须无毒无害,不得在生产用培养基内加入有毒有害物质和致敏性物质。有可能产生抗菌素、真菌毒素或其他活性物质的菌种还应包括有关抗菌素、真菌毒素或其他活性物质的检测报告。
(六)菌种的保藏方法、复壮方法及传代次数,防止菌种变异方法。
(七)对经过驯化、诱变的菌种,应提供驯化、诱变的方法及驯化剂、诱变剂等资料。
(八)生产的技术规范和技术保证。
(九)生产条件符合《保健食品生产良好规范》的证明文件。
(十)申请使用《可用于保健食品的真菌菌种名单》之外的真菌菌种研制、开发和生产保健食品,还应提供菌种具有功效作用的研究报告、相关文献资料和菌种及其代谢产物不产生任何有毒有害作用的资料。
第六条 申请人购买经过发酵或培养后的菌粉生产保健食品的,生产加工工艺只是混合、灌装过程,本规定第五条的资料也可由菌种原料供应商提供复印件(加盖原料供应商公章),并提供购销凭证。
第七条 样品试制单位应有专门的部门和人员管理生产菌种,建立菌种档案资料,内容包括菌种的来源、历史、筛选、检定、保存方法、数量、开启使用等完整的记录。
第八条 试制真菌类保健食品的场所应具备以下条件:
(一)符合《保健食品良好生产规范》(GMP)要求,并建立危害分析关键控制点(HACCP)质量保证体系。
(二)必须具备中试生产规模,即每日至少可生产500L的能力,并以中试产品报批。
(三)必须有专门的厂房或车间,有专用的生产设备和设施。必须配备真菌实验室,菌种必须有专人管理,应由具有中级以上技术职称的微生物专业的技术人员负责。制定相应的详细技术规范和技术保证。
第九条 生产用菌种及生产工艺不得变更。
第十条 凡是利用真菌菌丝体发酵生产的保健食品,如菌丝体容易获得子实体,可冠以其子实体加菌丝体命名;否则应以实际种名加菌丝体命名其产品(包括原料名称),不得冠以其子实体的名称。
第十一条 所用真菌菌种在其发酵过程中,除培养基外,不得加入具有功效成分的动植物及其它物质。
第十二条 经过基因修饰的菌种不得用于生产保健食品。
第十三条 本规定由国家食品药品监督管理局负责解释。
第十四条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
可用于保健食品的真菌菌种名单
酿酒酵母 Saccharomyces cerevisiae
产朊假丝酵母 Candida utilis
乳酸克鲁维酵母 Kluyveromyces lactis
卡氏酵母 Saccharomyces carlsbergensis
蝙蝠蛾拟青霉 Paecilomyces hepiali Chen et Dai, sp. Nov
蝙蝠蛾被毛孢 Hirsutella hepiali Chen et Shen
灵芝 Ganoderma lucidum
紫芝 Ganoderma sinensis
松杉灵芝 Ganoderma tsugae
红曲霉 Monacus anka
紫红曲霉 Monacus purpureus
益生菌类保健食品申报与审评规定(试行)
第一条 为规范益生菌类保健食品审评工作,确保益生菌类保健食品的食用安全,根据《中华人民共和国食品卫生法》、《保健食品注册管理办法(试行)》,制定本规定。
第二条 益生菌类保健食品系指能够促进肠道菌群生态平衡,对人体起有益作用的微生态产品。
第三条 益生菌菌种必须是人体正常菌群的成员,可利用其活菌、死菌及其代谢产物。
益生菌类保健食品必须安全可靠,即食用安全,无不良反应;生产用菌种的生物学、遗传学、功效学特性明确和稳定。
第四条 可用于保健食品的益生菌菌种名单由国家食品药品监督管理局公布。
第五条 国家食品药品监督管理局对保健食品的益生菌菌种鉴定单位进行确定,确定的菌种鉴定单位的名单由国家食品药品监督管理局公布。
益生菌类保健食品的菌种鉴定工作应在国家食品药品监督管理局确定的鉴定单位进行。
第六条 申请益生菌类保健食品,除按保健食品注册管理有关规定提交申报资料外,还应提供以下资料:
(一)产品配方及配方依据中应包括确定的菌种属名、种名及菌株号。菌种的属名、种名应有对应的拉丁学名。
(二)菌种的培养条件(培养基、培养温度等)。
(三)菌种来源及国内外安全食用资料。
(四)国家食品药品监督管理局确定的鉴定机构出具的菌种鉴定报告。
(五)菌种的安全性评价资料(包括毒力试验)。
(六)菌种的保藏方法。
(七)对经过驯化、诱变的菌种,应提供驯化、诱变的方法及驯化剂、诱变剂等资料。
(八)以死菌和/或其代谢产物为主要功能因子的保健食品应提供功能因子或特征成分的名称和检测方法。
(九)生产的技术规范和技术保证。
(十)生产条件符合《保健食品生产良好规范》的证明文件。
(十一)使用《可用于保健食品的益生菌菌种名单》之外的益生菌菌种的,还应当提供菌种具有功效作用的研究报告、相关文献资料和菌种及其代谢产物不产生任何有毒有害作用的资料。
第七条 申请人购买活菌种冻干粉直接生产保健食品,生产加工工艺只是混合、灌装过程,本规定第六条的资料也可由活菌种冻干粉原料供应商提供复印件(加盖原料供应商公章),并提供购销凭证。
第八条 申请注册的用于益生菌类保健食品生产的菌种应满足以下条件:
(一)保健食品生产用菌种应采用种子批系统。原始种子批应验明其记录、历史、来源和生物学特性。从原始种子批传代、扩增后保存的为主种子批。从主种子传代、扩增后保存的为工作种子批。工作种子批的生物学特性应与原始种子批一致,每批主种子批和工作种子批均应按规程要求保管、检定和使用。在适宜的培养基上主种子传代不超过10代,工作种子传代不超过5代。
(二)试制单位应有专门的部门和人员管理生产菌种,建立菌种档案资料,内容包括菌种的来源、历史、筛选、检定、保存方法、数量、开启使用等完整的记录。
(三)菌种及其代谢产物必须无毒无害,不得在生产用培养基内加入有毒有害物质和致敏性物质。
(四)从活菌类益生菌保健食品中应能分离出与报批和标识菌种一致的活菌。
第九条 益生菌类保健食品样品试制的场所应具备以下条件:
(一)符合《保健食品生产良好规范》(GMP)的要求,并建立危害分析关键控制点(HACCP)质量保证体系。
(二)具备中试生产规模,即每日至少可生产500L的能力,并以中试产品报批。
(三)必须有专门的厂房或车间、有专用的生产设备和设施;必须配备益生菌实验室,菌种必须有专人管理,应由具有中级以上技术职称的细菌专业的技术人员负责;制定相应的详细技术规范和技术保证。
第十条 生产用菌种及生产工艺不得变更。
第十一条 不提倡以液态形式生产益生菌类保健食品活菌产品。
第十二条 活菌类益生菌保健食品在其保质期内活菌数目不得少于106cfu/mL(g)。
第十三条 益生菌类保健食品如需在特殊条件下保存,应在标签和说明书中标示。
第十四条 所用益生菌菌种在其发酵过程中,除培养基外,不得加入具有功效成分的动植物及其它物质。
第十五条 经过基因修饰的菌种不得用于保健食品。
第十六条 本规定由国家食品药品监督管理局负责解释。
第十七条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
可用于保健食品的益生菌菌种名单
两岐双岐杆菌 Bifidobacterium bifidum
婴儿双岐杆菌 Bifidobacterium infantis
长双岐杆菌 Bifidobacterium longum
短双岐杆菌 Bifidobacterium breve
青春双岐杆菌 Bifidobacterium adolescentis
德氏乳杆菌保加利亚种 Lactobacillus delbrueckii subsp. bulgaricus
嗜酸乳杆菌 Lactobacillus acidophilus
干酪乳杆菌干酪亚种 Lactobacillus casei subsp. Casei
嗜热链球菌 Streptococcus thermophilus
罗伊氏乳杆菌 Lactobacillus reuteri
核酸类保健食品申报与审评规定(试行)
第一条 为规范核酸类保健食品审评工作,确保核酸类保健食品的食用安全,根据《中华人民共和国食品卫生法》和《保健食品注册管理办法(试行)》,制定本规定。
第二条 核酸类保健食品系指以核酸(DNA或RNA)为原料,辅以相应的协调物质生产的保健食品。
第三条 申请核酸类保健食品,除须按保健食品的要求提交资料外,还应当提供以下资料:
(一)产品配方及配方依据中应明确所用核酸的具体成分名称、来源、含量;
(二)与所申报功能直接相关的科学文献依据;
(三)企业标准中应明确标出所用核酸各成分的含量、纯度和相应的定性、定量检测方法以及质量标准;
(四)提供所用核酸原料的详细生产工艺(包括加工助剂名称、用量);
(五)国家食品药品监督管理局确定的检验机构出具的核酸原料的纯度检测报告。
第四条 不得以单一的DNA或RNA作为原料申报保健食品。
第五条 保健食品中所使用核酸,其单一原料纯度应大于80%。
第六条 核酸类保健食品的功能申报范围暂限定为增强免疫力功能。
第七条 核酸类保健食品按照保健食品功能学评价程序和方法进行保健功能学评价试验时,除按推荐摄入量规定倍数设立高、中、低三个剂量组,还需增设中剂量配料对照组(产品除核酸外的所有其他配料),当样品组与空白对照组、配料组比较均有统计学差异时,该产品方可以核酸作为功效成分进行标注。
第八条 核酸类保健食品产品说明书中功效成分一项,应当根据国家食品药品监督管理局确定的检验机构出具的检测报告的实测值,明确标出产品中具体核酸成分的含量。
第九条 核酸类保健食品,其核酸的每日推荐食用量为0.6g-1.2g。
第十条 所有保健食品均不得以“核酸”命名。
第十一条 核酸类保健食品说明书及标签中的“不适宜人群”除按保健食品相关规定标注外,应明确标注出“痛风患者”。
第十二条 本规定由国家食品药品监督管理局负责解释。
第十三条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
野生动植物类保健食品申报与审评规定(试行)
第一条 为保护野生动植物,规范野生动植物类保健食品申报与审评工作,根据《中华人民共和国野生动物保护法》、《中华人民共和国野生植物保护条例》、《保健食品注册管理办法(试行)》,制定本规定。
第二条 野生动植物类保健食品是指使用了国务院及其农业(渔业)、林业行政主管部门发布的国家保护的野生动物、植物名录中收入的野生动物、植物品种生产的保健食品。
第三条 禁止使用国家一级和二级保护野生动植物及其产品作为保健食品原料。
第四条 禁止使用人工驯养繁殖或人工栽培的国家一级保护野生动植物及其产品作为保健食品原料。使用人工驯养繁殖或人工栽培的国家二级保护野生动植物及其产品作为保健食品原料的,应提供省级以上农业(渔业)、林业行政主管部门出具的允许开发利用的证明文件。
第五条 使用国家保护的有益的或者有重要经济、科学研究价值的陆生野生动植物及其产品作为保健食品原料的,应提供省级以上农业(渔业)、林业行政主管部门依据管理职能出具的允许开发利用的证明文件。
第六条 使用中华人民共和国林业植物新品种保护名录中植物及其产品作为保健食品原料的,如果该种植物已获“品种权”,应提供该种植物品种权所有人许可使用的证明;如该种植物尚未取得品种权,应提供国务院林业主管部门出具的该种品种尚未取得品种权的证明。
第七条 对于进口保健食品中使用《濒危野生动植物种国际贸易公约》名录中动植物及其产品的,应提供国务院农业(渔业)、林业行政主管部门准许其进口的批准证明文件、进出口许可证及海关的证明文件。
第八条 禁止使用野生甘草、苁蓉和雪莲及其产品作为原料生产保健食品。使用人工栽培的甘草、苁蓉和雪莲及其产品作为保健食品原料的,应提供原料来源、购销合同以及原料供应商出具的收购许可证(复印件)。
第九条 本规定由国家食品药品监督管理局负责解释。
第十条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
氨基酸螯合物等保健食品申报与审评规定(试行)
第一条 为规范保健食品审评工作,根据《中华人民共和国食品卫生法》和《保健食品注册管理办法(试行)》,制定本规定。
第二条 申请注册使用氨基酸螯合物生产的保健食品,除按保健食品注册管理有关规定提交有关资料外,还应提供如下资料:
(一)提供明确的产品化学结构式、物理化学性质,配体与金属离子之比、游离元素和总元素之比。
(二)提供氨基酸螯合物定性、定量的检测方法(包括原料和产品)以及国家食品药品监督管理局确定的检验机构出具的验证报告。
(三)国家食品药品监督管理局确定的检验机构出具的急性毒性试验加做停食16小时后空腹一次灌胃试验(分别在灌胃2小时、4小时后重点观察消化道大体解剖和病理变化情况)和30天喂养试验[肝、肾、胃肠(包括十二指肠、空肠、回肠)]的组织病理报告。
(四)国内外该氨基酸螯合物食用的文献资料。
第三条 申请注册使用微生物发酵直接生产的保健食品,除按保健食品有关规定提交相关资料外,还需提供下列资料:
(一)菌种来源及国家食品药品监督管理局确定的检验机构出具的菌种鉴定报告。
(二)菌种的毒力试验报告。
(三)菌种的安全性评价报告。
(四)国内外该菌种用于食品生产的文献资料。
(五)发酵终产物的质量标准(包括纯度、杂质成分及含量)。
第四条 申请注册以褪黑素为原料生产的保健食品,除按照保健食品注册有关规定提交资料外,还需提供下列资料,并符合下列要求:
(一)产品配方中除褪黑素和必要的辅料(赋形剂)外,不得添加其他成分(维生素B6除外)。
(二)申请人应提供褪黑素原料的检测报告,其纯度应达到99.5%以上。
(三)褪黑素的推荐食用量为1~3mg/日。
(四)申报的保健功能暂限定为改善睡眠。
(五)注意事项中应注明从事驾驶、机械作业或危险操作者,不要在操作前或操作中食用和自身免疫症(类风湿等)及甲亢患者慎用。
第五条 申请注册以大豆磷脂为原料生产的保健食品,除按照保健食品注册有关规定提交资料外,还需提供下列资料,并符合下列要求:
(一)申请人应提供大豆磷脂原料的丙酮不溶物和乙醚不溶物含量检测报告。
(二)使用的大豆磷脂原料应符合《磷脂通用技术条件》(SB/T10206)中一级品的要求。
第六条 申请注册以芦荟为原料生产的保健食品,除按照保健食品注册有关规定提交资料外,还需提供下列资料,并符合下列要求:
(一)申请人须提供省级以上专业鉴定机构出具的芦荟品种鉴定报告。
(二)可作为保健食品原料的芦荟品种为库拉索芦荟和好望角芦荟。其他芦荟品种应按有关规定,提供该品种原料的安全性毒理学评价试验报告及相关的食用安全的文献资料。
(三)芦荟的食用量控制在每日2g以下(以原料干品计)。以芦荟凝胶为原料的除外。
(四)芦荟原料应符合《食用芦荟制品》(QB/T2489)的要求。
(五)不适宜人群须标明孕产妇、乳母及慢性腹泻者。
(六)注意事项须注明食用本品后如出现明显腹泻者,请立即停止食用。
第七条 申请注册以蚂蚁为原料生产的保健食品,除按照保健食品注册有关规定提交资料外,还需提供下列资料,并符合下列要求:
(一)申请人应提供省级以上专业鉴定机构出具的蚁种鉴定报告,并需提供蚂蚁原料来源证明。
(二)可作为保健食品原料的蚂蚁品种为拟黑多刺蚁、双齿多刺蚁、黑翅土白蚁、黄翅大白蚁、台湾乳白蚁。其他蚂蚁品种应按有关规定,提供该品种原料的安全性毒理学评价试验报告及相关的食用安全的文献资料。
(三)产品生产加工过程中,温度一般不超过80℃。
(四)提供蚁酸含量测定报告。
(五)注意事项须注明过敏体质者慎用。
第八条 申请注册以酒为载体的保健食品,除按照保健食品注册有关规定提交资料外,还需提供下列资料,并符合下列要求:
(一)产品酒精度数不超过38度。
(二)每日食用量不超过100ml。
(三)不得申报辅助降血脂和对化学性肝损伤有辅助保护功能。
第九条 申请注册不饱和脂肪酸类保健食品应符合下列要求:
(一)产品的每日推荐食用量不超过20ml。
(二)食用方法不得加热烹调。
(三)产品以每日食用量定量包装。
第十条 申请注册以甲壳素为原料生产的保健食品,除按照保健食品注册有关规定提交资料外,还需提供下列资料,并符合下列要求:
(一)申请人应提供甲壳素原料的脱乙酰度检测报告。
(二)甲壳素原料的脱乙酰度应大于85%。
第十一条 申请注册以超氧化物歧化酶(SOD)为原料生产的保健食品应符合下列要求:
(一)超氧化物歧化酶(SOD)应从天然食品的可食部分提取,其提取加工过程符合食品生产加工要求。
(二)以超氧化物歧化酶(SOD)为原料生产的保健食品,申报的保健功能暂限定为抗氧化。
(三)以超氧化物歧化酶(SOD)单一原料申请保健食品时,应提供超氧化物歧化酶(SOD)在人体内口服吸收利用率、体内代谢等的国内外研究资料,证明超氧化物歧化酶(SOD)可经口服吸收。
(四)以超氧化物歧化酶(SOD)组合其他功能原料申请保健食品时,加入的功能原料应具有抗氧化作用。产品不得以超氧化物歧化酶(SOD)命名,不得宣传超氧化物歧化酶(SOD)的作用。
第十二条 申请注册以下原料生产的保健食品,除按保健食品规定提交申报资料外,还应提供:
(一)使用动物性原料(包括胎盘、骨等)的,应提供原料来源证明及县级以上畜牧检疫机构出具的检疫证明。
(二)使用红景天、花粉、螺旋藻等有不同品种植物原料的,应提供省级以上专业鉴定机构出具的品种鉴定报告。
(三)使用石斛的,应提供省级以上专业鉴定机构出具的石斛品种鉴定报告和省级食品药品监督管理部门出具的人工栽培现场考察报告。
第十三条 本规定由国家食品药品监督管理局负责解释。
第十四条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
应用大孔吸附树脂分离纯化工艺生产的保健食品申报与审评规定(试行)
第一条 为规范应用大孔吸附树脂分离纯化工艺生产的保健食品审评工作,确保保健食品的食用安全,根据《中华人民共和国食品卫生法》和《保健食品注册管理办法(试行)》,制定本规定。
第二条 应用大孔吸附树脂分离纯化工艺生产的保健食品是指产品生产过程中及原料生产过程中应用了大孔吸附树脂分离纯化工艺的保健食品。
第三条 申请应用大孔吸附树脂分离纯化工艺生产的保健食品除按照保健食品注册管理的有关规定提交资料外,还应提供以下资料:
(一)大孔吸附树脂的相关资料
1、大孔吸附树脂规格标准。标准内容应包括大孔吸附树脂名称、牌(型)号、结构、合成原料(主要原料、交联剂、致孔剂、分散剂等名称和规格)、外观、极性和粒径范围、含水量、湿密度、干密度、比表面积、孔径、孔隙率、孔容等,并提供大孔吸附树脂标准级别等。
2、大孔吸附树脂使用说明书。使用说明书的内容应包括:
(1)大孔吸附树脂性能简介、适用范围、主要原料和添加剂种类与名称;
(2)残留物(包括未聚单体、交联剂、主要添加剂)及其残留量检测方法和限量标准及依据;
(3)使用方法和注意事项,包括新大孔吸附树脂的预处理方法、再生处理方法和操作注意事项、贮存条件等,以及可能出现异常情况的处理方法。
3、生产批号、生产时间、产品检验报告书。
4、相关证明文件。大孔吸附树脂生产企业的企业名称、地址、电话、营业执照及相关生产许可证件的复印件等。
(二)应用大孔吸附树脂进行分离纯化的制备工艺研究资料
1、制备工艺中应用大孔吸附树脂进行分离纯化的目的与依据。详细说明应用大孔吸附树脂进行分离、纯化的目的和必要性,并提供相关研究或文献资料。
2、大孔吸附树脂的预处理方法和合格标准。预处理方法包括考察预处理溶剂的种类、用量、浸泡时间、流速、温度、pH值等工艺参数和操作规程。
3、生产工艺的研究资料。大孔吸附树脂型号的选择、比上柱量、比吸附量、比洗脱量、树脂柱的径-高比、提取液的适宜上柱温度、pH及流速、解吸附溶剂及其条件的选择、解吸附终点判定方法等研究资料,并将上述资料作为该产品的生产工艺的一部分。
4、大孔吸附树脂再生方法的确定。大孔吸附树脂经使用后,吸附能力下降,应进行再生处理。根据功效成分和大孔吸附树脂的理化性质,制订大孔吸附树脂再生处理方法及其合格标准,申请人应制定相应的标准、操作规程并列入企业标准的附录。
(三)使用以大孔吸附树脂分离纯化工艺制造的原料生产的保健食品,申请时还应提供原料生产企业的详细资料,原料的制备工艺和详细的质量标准,包括原料中大孔吸附树脂残留物的标准和检测报告。申请人应提供由国家食品药品监督管理局确定的检验机构出具的该原料大孔吸附树脂残留物的检验报告。
(四)大孔吸附树脂及其纯化工艺等安全性评价资料
第四条应用苯乙烯骨架型树脂分离纯化工艺的保健食品应符合以下要求:
(一)大孔吸附树脂应用前应进行预处理,预处理以后的大孔吸附树脂中的有机残留物应控制在安全范围内,对苯乙烯骨架型树脂要求苯的残留量小于2mg/kg、二乙烯苯小于20 mg/kg。对其它类型的大孔吸附树脂,根据具体情况确定限量标准。
(二)一般情况下,不得使用再生后的比吸附量仍下降达30%以上时的大孔吸附树脂。
(三)应用以大孔吸附树脂分离纯化工艺生产保健食品原料的生产企业,应当对原料的大孔吸附树脂残留物进行检验或复核。对用苯乙烯骨架型树脂制备的原料中二乙烯苯含量要求为小于50μg/kg,并将此列入企业标准。如果原料质量达到上述标准的,可以免测该保健食品终产品的大孔吸附树脂残留物的含量。
(四)建立树脂残留物的检测方法和标准,并列入企业标准。
(五)对保健食品生产过程中应用苯乙烯骨架型树脂分离纯化工艺的保健食品,其产品中二乙烯苯含量要求为小于50 μg/kg。
第五条 其它类型的大孔吸附树脂参照苯乙烯骨架型并结合具体品种制定相应的残留物及残留标准等内容。
第六条 原则上不得以多个动植物原料混合后再应用大孔吸附树脂纯化工艺生产保健食品。
第七条 必要时,国家食品药品监督管理局可对大孔吸附树脂生产企业和应用大孔吸附树脂生产保健食品所用原料的企业进行现场核查。
第八条 本规定由国家食品药品监督管理局负责解释。
第九条 本规定自二○○五年七月一日起实施。以往发布有关规定与本规定不一致的,以本规定为准。
保健食品申报与审评补充规定(试行)
第一条 为规范保健食品审评工作,根据《中华人民共和国食品卫生法》和《保健食品注册管理办法(试行)》,制定本规定。
第二条 同一产品原则上不得制成两种剂型或分别成型,符合下列条件的产品,可申请注册两种剂型或分别成型:
(一)有足够的证据说明,目前的生产技术条件下,不能加工成一种剂型;或虽可加工成一种剂型,但影响产品保质期,其保质期小于6个月的;
(二)单独的一种剂型,不能独立形成一种保健功能。
(三)两种剂型的保健食品其产品名称应标明两种剂型,产品的最小包装应为一日或一次食用量。
第三条 以舌下吸收的剂型、喷雾剂等不得作为保健食品剂型。
第四条 缓释制剂保健食品审评的具体规定为:
(一)申请人应提供充分的证据说明缓释制剂的必要性(包括文献及试验依据)。
(二)产品的原料构成应为单体成分,其纯度为90%以上。
(三)应保证普通制剂改为缓释制剂产品的食用安全,提交相关的安全性毒理学评价资料。
(四)申请人应提供按照《中华人民共和国药典》中《缓释控释制剂指导原则》、《释放度测定方法》、《药物制剂人体生物利用度和生物等效性试验指导原则》进行普通制剂和缓释制剂的释放度比较试验、人体生物利用度和生物等效性试验的试验报告。
第五条 保健食品原料与主要辅料相同,涉及不同口味、不同颜色的产品,可免做安全性毒理学评价试验和功能学评价试验(提供原产品试验报告的复印件),但其检验方法、评价指标和判断标准应符合现行的规定。在产品名称后注明口味或颜色。
第六条 增补剂型的产品,申请人应说明增补剂型的必要性和依据。其产品原料、功效成分、每日食用量应与原产品相同,产品的性状相似,如同为固体或同为液体,并且产品的生产工艺无质的变化,不影响安全和功能,可免做安全性毒理学评价试验和功能学评价试验(提供原产品试验报告的复印件),但其检验方法、评价指标和判断标准应符合现行的规定。
第七条 不得以肌酸和熊胆粉作为原料申请保健食品,暂不受理和审批以金属硫蛋白为原料申请的保健食品。
第八条 保健食品中使用的辅料一般应为《食品添加剂使用卫生标准》或卫生部公告的食品添加剂新品种名单中的品种,否则,应当提供该辅料食用安全及国内外使用情况的相关资料。
第九条 保健食品的适宜人群、不适宜人群、注意事项应根据申报的保健功能和产品的特性确定。
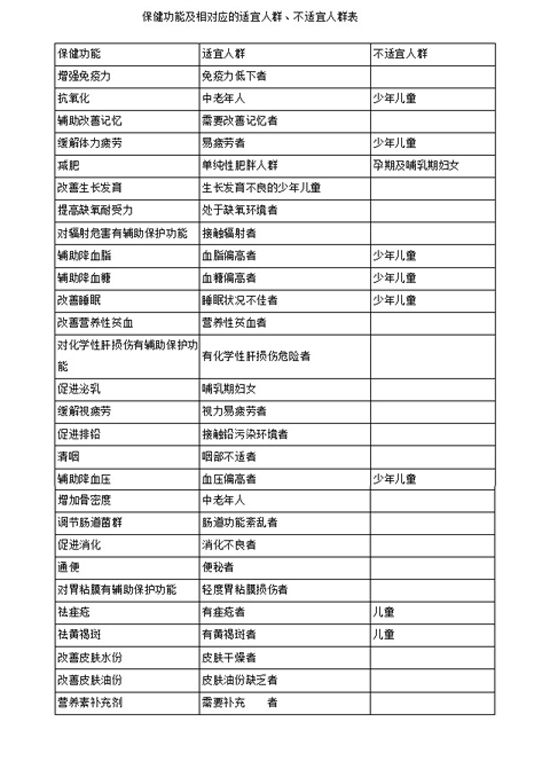
(一)保健功能及相对应的适宜人群、不适宜人群应当参照《保健功能及相对应的适宜人群、不适宜人群表》的要求。一个保健食品具有两个以上保健功能的,以主要功能的适宜人群和不适宜人群为主,综合确定产品的适宜人群和不适宜人群。
(二)产品中使用的某种原料不适宜特定人群食用的,产品的不适宜人群应增加该特定人群。例如:使用红花为原料的,其产品不适宜人群应增加孕产妇、月经过多者;使用灵芝、人参、西洋参及含激素的物品(如蜂皇浆)等为原料的保健食品,不适宜人群应增加少年儿童。
第十条 本规定由国家食品药品监督管理局负责解释。
第十一条 本规定自二○○五年七月一日起实施。以往发布的规定,与本规定不符的,以本规定为准。
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


