

药品工艺变更频繁爆雷的背后,实际上是在企业运行以及监管机制方面都出现了问题。一方面,强大的产业一定与强大的监管互为支撑,将监管嵌入产业势在必行。而另一方面,采用更合理以及更能落地的监管政策,无疑才是解决药品工艺更改这一行业沉疴的关键路径。
步入2018年以来,因为药品工艺变更而被点爆的雷频繁出现,最典型的如长春长生。这场因为随意变更工艺参数和设备而使全行业为之震动的问题疫苗事件,其背后正是药品生产工艺随意变更而为药品质量安全带来的重大隐患。这让整个行业的神经再次绷紧。
无论是从企业自身角度来讲还是从国家监管层面出发,使药品生产工艺变更的管理更规范化,防止更严重的药品安全事件爆发,已经迫在眉睫,必须引起足够重视。
单从监管层面来看,监管机构并非没有意识到药品工艺变更给药品安全所带来的巨大隐患。2016年8月,原国家食品药品监督管理总局(以下简称“原食药监总局”)发布《关于开展药品生产工艺核对工作的公告(征求意见稿)》,将新一轮的工艺核查风暴正式拉开。文件的内容一针见血:仍有部分2007年前批准上市的品种,未按照批准的生产工艺组织生产、改变生产工艺不按规定研究和申报。
而据中国医药企业管理协会此前组织行业近百家企业所进行的问卷调研和实际访谈显示,86.21%的企业的2007年前批准上市品种曾进行过工艺变更,而只有22.41%的企业选择了进行补充申报。
3月4日下午,由中国医药行业25家协(学)会共同主办的2019“声音·责任”医药界人大代表政协委员座谈会将于北京正式举行,而通过之前的调研集合行业专家意见提供的三份主提案中,其中一份就是关于药品生产工艺核对和工艺变更管理的相关建议,并对此针对性提出了五项具体解决办法。
1
超八成企业生产工艺变更未申报
为准确了解中国医药行业企业生产工艺变更的真实状态,此前,中国医药企业管理协会组织对广东、浙江、辽宁、重庆等省市中药、化学药、生物药、原料药等近百家相关企业进行了问卷调研和实际访谈。结果显示,绝大部分药企都进行过生产工艺变更,且大部分企业未完成工艺变更补充申请。
调查要求企业对2007年前获批的品种,按照存在生产工艺变更情况产品占所有品种的比例划分了6个档次。结果发现,仅13.79%的企业无生产工艺变更情况。也就是说,在样本中超过八成的药企或多或少都进行过生产工艺变更。
2007年获批品种中变更生产工艺的比例
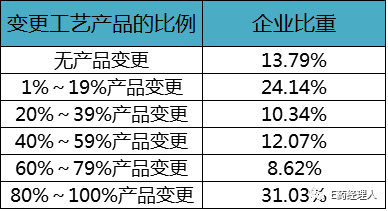
再进一步对工艺变更后是否进行补充申请的情况调研,结果显示,只有22.41%的企业选择完成了补充申请,而剩下的企业都存在未按照规定完成变更生产工艺补充申请的情况。
那么企业为何不按规定进行补充申请?受访企业归纳出三个主要原因:
一是企业与药监部门对微小变更、中等变更、重大变更的理解不同;在实际操作中,由于生产工艺包括的内容很多且情况复杂,致使企业与药监部门对变更的归类理解不一致,从而有的企业不知道实际发生的工艺变更应提交补充申请还是只需要内部备案。超过62%的企业都存在这方面的原因。
二是完成生产工艺变更的补充申请需耗费大量资金与时间成本;三是完成生产工艺变更的补充申请流程长、时间久。同样有50%到60%的企业困扰于此。
除此之外,还存在诸多原因导致企业不愿意主动进行生产工艺变更的申报,例如出于对泄露自家工艺秘密的担心,以及获批药品长期未放大生产等,这些原因分别也占到了27.59%和24.14%的比例。
2
生产工艺变更为何难规范管理?
实际上,国家层面对于药品生产工艺变更的关注由来已久并且很早就开始有所行动。
早在2007年和2009年,监管部门分别对注射剂和基本药物进行了工艺核查;2016年8月则是原食药监总局的新一轮工艺核查风暴,到2017年,原食药监总局又连发《已上市化学药品生产工艺变更研究技术指导原则》和《已上市中药生产工艺变更研究技术指导原则》两文,指导企业对已上市药品拟变更生产工艺开展研究。
但一直以来,药品工艺变更大多不报备的情况仍然显著,更重要的是,因生产工艺变更导致的药品质量安全事件愈发严重。
究其原因,首先是因为存在众多历史遗留问题。回溯药品监管的历史,在2001年以前国家的药品注册分散在各个省市区,因而或多或少存在标准不一的情况;到了2000年~2006年期间,出现了药品注册报批数量飙升的现象,申报资料乱象纷呈,甚至存在倒卖资料。因此当时很大一部分申报的工艺用于实际生产时会有出入,企业为了生产出合格的药品不得不更改生产工艺。
另外,有的药企为了工艺的保密,在申报时会作出虚假工艺陈述,或者故意提交不完整或参数模糊的注册工艺。
随着科技发展和产业升级,现实条件下药品的生产工艺会因很多客观因素而更改。在中国医药企业管理协会组织的调查中,技术革新、药典等各类标准的提升、生产场所及生产设备改变、适应放大生产等成为药企变更生产工艺的几个最主要的原因。
而从现实监管层面来看,的确也存在很多挑战。
有行业专家认为,中国现在有16.7万个药品批准文号,但许多原始申报工艺已经丢失,企业自行变更生产工艺的问题非常普遍,监管部门难以“摸清底数”。因此,当前并不是大多数企业都能提供产品批准生产时的生产工艺资料,很多企业只能提供部分产品批准工艺资料甚至无法提供。
事实上,据中国医药企业管理协会的调研统计显示,关于批准工艺的完整性,实际上只有36.21%的企业能够提供产品批准生产时的生产工艺资料,超过一半的企业只能提供部分产品批准工艺资料。而5.17%的企业则完全无法提供相关资料。
还有专家表示,监管缺乏震慑力、日常监管技术滞后也或多或少影响药品生产工艺变更的规范管理。他指出,在欧美等发达国家,监管部门已使用数字化手段等新技术对药品的生产流程进行监管,造假或工艺违规操作等不良行为能有效被发现和惩治。
3
如何规范监管?
由于绝大多数制药企业都不同程度地对原有工艺进行了调整更改,但由于报批报备制度滞后繁琐,使得很多企业没有履行变更程序。但毋庸置疑的是,这一影响药品质量安全的重大隐患急需根除。因此行业协会联合提出如下建议:
首先,细化明确不同变更类别的内涵。建议指出按照已上市药品技术变更指导原则,将变更分为三类,且分别有不同的管理要求。
因为在实际操作中,由于生产工艺内容多且杂,致使企业与药监部门对变更的归类理解不一致,从而有的企业不知道实际发生的工艺变更应提交补充申请还是只需要内部备案。因此需要组织专家进一步细化并明确不同变更类别的内涵,指导地方和企业开展相关工作。
第二,分阶段开展工艺变更自查核查工作。药品生产工艺变更的自查核查和整改涉及到大部分企业,对不同类型的产品,比如口服固体制剂、中成药、注射剂、血液制品、疫苗等,按照一定的标准进行分类,分阶段完成。另外发挥生产监督(GMP检查)的作用,特别是对非重大变更,在GMP检查中增加对该部分的检查力度,关注非重大变更的自行评估和资料完整性。
第三,分层次完成补充申请申报工作。为了提升药品生产工艺自查核查的效率,降低时间成本,建议针对药品生产工艺自查核查的补充申请,I类变更企业内备案,II类变更补充申请提交地方药监局,III类变更除部分特殊产品,如注射剂、血液制品、生物制品等,需提交国家药监局审评审批外,其余只要提交地方药监局即可。
第四,尊重现实,允许企业根据现有工艺补充调整工艺档案。相当数量的企业原始申报的工艺不完整、不准确,后续变更也未及时申报,面对这些现实问题,建议本着实事求是的原则,允许企业根据现有生产工艺补充调整工艺档案,并重点核查企业现有工艺的稳定性和可靠性。
另外做好企业工艺保密工作,落实《关于加强药品审评审批信息保密管理的实施细则》,建立药监部门对企业生产工艺资料的保密机制,打消企业在申报工艺时的顾虑。同时,借鉴国外DMF管理制度,建立原料药工艺技术信息分层管理制度,允许制剂采购商能够部分获取原料药生产技术信息。
关于举办中药饮片炮制技术研究与质量控
各会员单位: 为顺应药品监管规..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


