

2025年9月3日,FDA正式推出"罕见病证据原则"(Rare Disease Evidence Principles, RDEP)加速审评通道,旨在提升针对超罕见遗传病疗法的审评效率。
申办方可在关键性临床试验启动前随时申请RDEP,但需满足以下条件:
(1)适应症须为美国境内不足1000人的超级罕见病;
(2)治疗对象为明确的先天性遗传缺陷,该缺陷是病理机制的主要驱动因素;
(3)疾病进程快速恶化,会在短时间内导致严重功能障碍或死亡;
(4)当前无有效替代治疗方案。
众所周知,传统多中心临床试验模式在罕见病领域面临现实挑战。具体而言包括两大难点:一是患者少,二是伦理上不允许设置安慰组。这种情况下,企业通常需要通过基于真实世界研究的单臂临床试验(Single Arm Trial,SAT)来帮助药物注册上市,但对如何设计临床试验和其他细节问题并不清晰。
对此,FDA药品审评与研究中心(CDER)与生物制品审评与研究中心(CBER)联合制定了RDEP方案,通过纳入多元证据类型来平衡科学严谨性与可行性。具体而言,新药审批可基于一项充分对照的临床试验(可能是SAT),并结合以下补充证据:
强有力的机制研究或生物标志物证据;
非临床模型研究数据;
临床药效学数据;
病例报告、扩大用药项目数据或疾病自然史研究。
为了符合条件,药企需通过正式会议请求提交申请材料。FDA鼓励患者组织和领域专家全程参与证据体系构建。值得一提的是,该流程与孤儿药资格认定《FD&C法案526条款》相互独立,申请RDEP审评并不自动获得孤儿药资格,申办方仍需按既定程序申请孤儿药认定。此外,通过经该通道获批的药品可能面临更严格的上市后监管要求。
对此,FDA局长Marty Makary博士指出:"这项原则确保了我们在现行法规框架下采取灵活务实的审评策略,通过多维度证据体系为申办方构建严谨、明确的研发路径,让最需要创新疗法的患者能够及时受益。"
实际上,Makary很早就关注到了罕见病的获批问题,他在2023年发表的研究中指出,2022年美国仅有12%的罕见病药物通过单臂试验获批,远低于欧盟的28% 。今年4月份(Makary上任不久),他在接受采访时就曾透露,FDA正在探索一条针对罕见病药物的 “新路径”。当时受此消息影响,多家罕见病药企股价上涨:Lexeo Therapeutics股价暴涨18.6%、Dyne Therapeutics股价暴涨16.3%、Regenxbio股价暴涨12.5%……
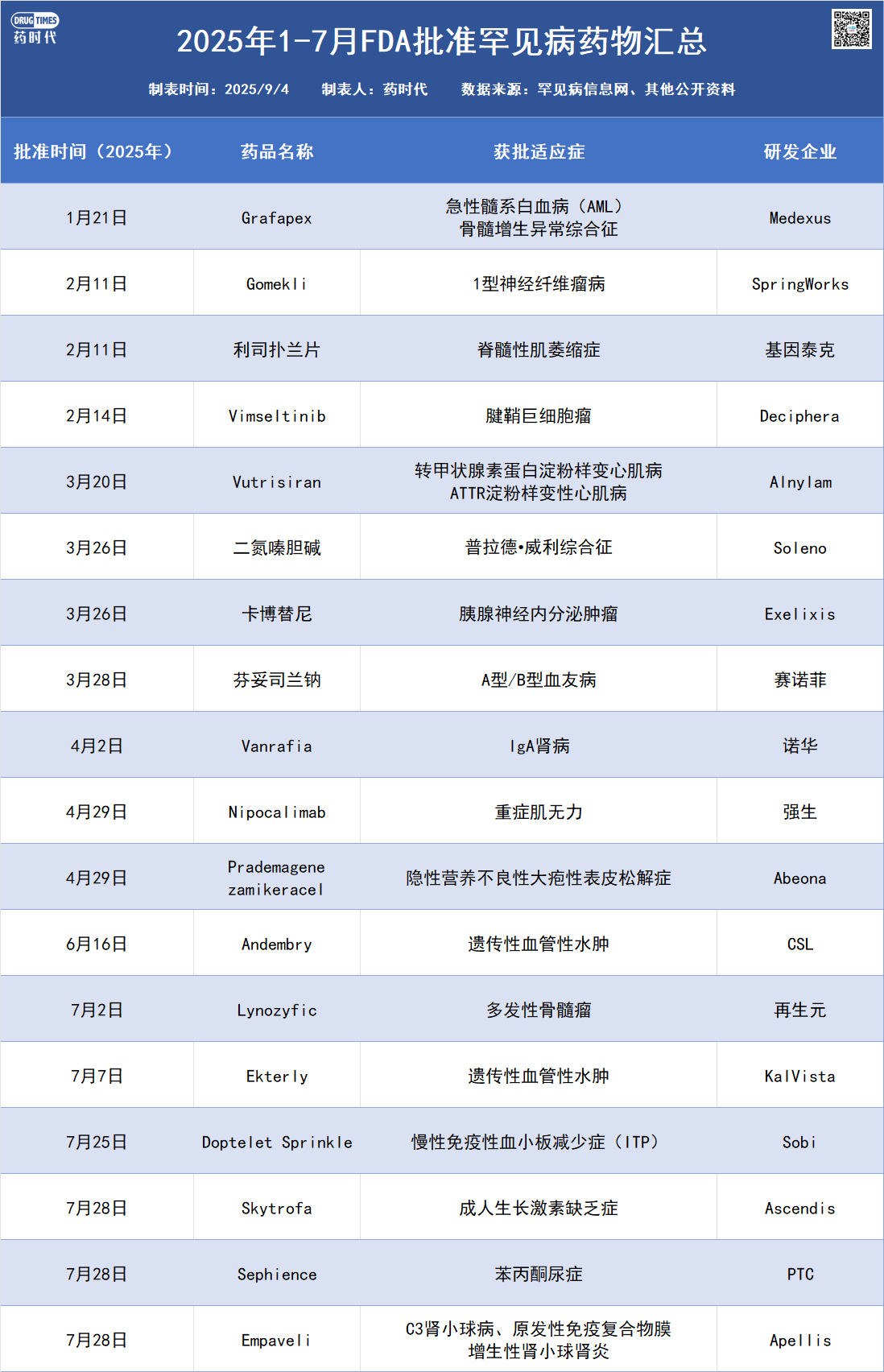
经罕见病信息网统计,2025年1-7月,FDA批准了18款罕见病药物。相信随着RDEP的推出,未来将有更多罕见病药物获批上市。不过也有业内人士持谨慎态度,William Blair分析师Sami Corwin在9月3日研报中指出,虽然RDEP的出台对罕见病药企是利好消息,但由于患者规模限制,使多数在研疗法难以适用。他列举了Neurogene、Rocket、Lexeo、Ultragenyx等公司为例,指出其研发项目早已采用单臂试验,但大多数无法满足RDEP条件,以此缩短审批周期。
参考资料:
1.FDA官网
2.UPDATE: Analyst questions impact of FDA's rare disease pathway proposal(fiercebiotech)
3.上半年,美国批准了18个罕见病药物!(附获批清单)(罕见病信息网)
4.其他公开资料
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


