

据 Insight 数据库统计,上周(5 月 26 日—6 月 1 日)全球共有 44 款创新药(含改良新)研发进度推进到了新阶段,其中 2 款获批上市,6 款获批临床,4 款申报临床。
本周末是 2024 ASCO 正式开幕的时间。无论国内国外,新药进度相关的审评动态都相对减少,取而代之的是精彩纷呈的临床结果、医药交易等。
下文,Insight 将分别摘取国内外部分重点项目做介绍。
国内创新药进展
国内部分,本周是新药获批的高峰。进度来看,共有 28 款创新药(含改良新)研发进度推进到了新阶段,除获批之外还有获批临床 3 款,申报临床 6 款。
本周国内首次启动临床的 6 款创新药(含改良新)
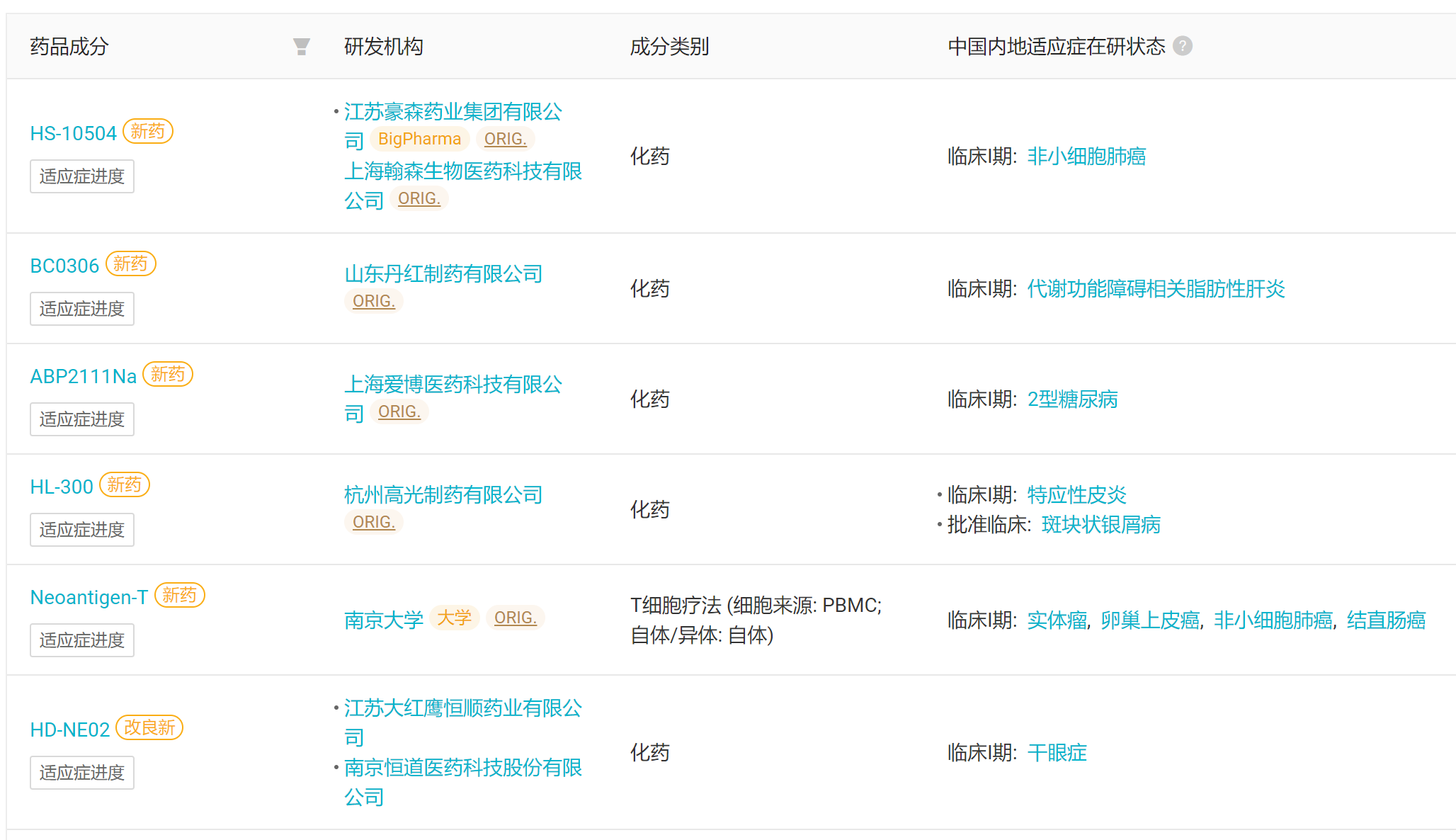
来自:Insight 数据库网页版
重磅临床结果
1、康方生物:「依沃西单抗」头对头 K 药 III 期临床成功,一线治疗 NSCLC
5 月 31 日凌晨 2 点,康方宣布,PD-1/VEGF 双抗「依沃西」单药对比帕博利珠单抗一线治疗 PD-L1 表达阳性局部晚期或转移性 NSCLC 的注册性 III 期临床研究 HARMONi-2,由独立数据监察委员会(IDMC)进行的预先设定的期中分析显示强阳性结果:达到无进展生存期(PFS)的主要研究终点。
此结果一出,曾以 50 亿美元与康方合作拿下该药的 Summit 公司股价立即狂涨 270% 以上,康方生物在翌日开盘后亦是强势拉升超 50%,一扫上周暴跌 40% 的低迷。
AK112-303(CTR20222137) 是一项评估依沃西(商品名:依达方®)单药对比帕博利珠单药一线治疗 PD-L1 表达阳性(PD-L1 TPS ≥ 1%)的局部晚期或转移性非小细胞肺癌(NSCLC)的注册性 III 期随机、双盲临床试验,主要研究终点为无进展生存期(PFS),共入组 398 例受试者,其中 PD-L1 TPS 1-49% 的受试者占比为 57.8%,PD-L1 TPS ≥ 50% 的受试者占比为 42.2%,与真实世界患者表达水平分布一致。
结果显示,在意向治疗人群 (ITT) 中,依沃西组相较于帕博利珠组显著延长了患者无进展生存期(PFS),风险比(HR)显著优于预期。依沃西组在 PD-L1 TPS 1-49% 和 PD-L1 TPS ≥ 50% 的人群中,PFS 获益均非常显著;依沃西组各个亚组疗效分析均显示强阳性结果,包括鳞癌、非鳞癌,有/无肝转移、有/无脑转移等患者人群;安全性方面,依沃西组总体安全性良好,无新的安全性信号。
HARMONi-2(AK112-303)临床研究详细数据仍待披露,据康方表示,将在即将举行的全球学术会议上公布。
依沃西由此成为了全球首个且唯一在 III 期单药头对头临床研究中证明疗效显著优于帕博利珠单抗的药物。单药展现的优越疗效和安全性,进一步夯实了该药作为肿瘤免疫治疗(IO)基石产品的巨大潜力,包括与 ADC 药物或其它新型抗癌药物联用的广阔临床开发价值和市场前景。
依沃西是全球第一个获批上市的「免疫治疗+抗血管生成」协同机制的肿瘤治疗双特异性抗体新药,可以同时靶向 PD-1 和 VEGF 靶点。目前,依沃西在全球范围内已经获批 1 项适应症,有 5 项 III 期临床研究正在开展,其中的 2 项国际多中心 III 期临床在海外开展,4 项为以 PD-1 单抗为阳性对照药物的 III 期临床研究。
在本届 ASCO 上,康方也以口头汇报形式公布了依沃西联合化疗用于经 EGFR-TKI 治疗进展的 EGFR 突变局部晚期或转移性 nsq-NSCLC Ⅲ 期临床研究 HARMONi-A 的期中分析成果,详情可见企业报道:ASCO Oral & JAMA 主刊丨Ⅲ 期 HARMONi-A 研究重磅公布。
2、科伦博泰:TROP-2 ADC 强势出击,三阴乳腺癌、非小细胞肺癌两大临床结果披露
5 月 31 日,2024 年度 ASCO 大会开幕。
在本次会议中,科伦药业在 5 月 31 日下午的口头报告环节披露了芦康沙妥珠单抗 (sac-TMT,研发代号:SKB264/MK-2870) 联合 KL-A167(抗 PD-L1 单抗)用于一线治疗晚期 NSCLC 患者的 2 期研究 (OptiTROP-Lung01) 结果,摘要编号 #8502;
在 6 月 2 日上午,又报告了芦康沙妥珠单抗用于既往接受过治疗的局部复发或转移性三阴性乳腺癌 (TNBC) 患者的 3 期研究 (OptiTROP-Breast01) 结果,摘要编号 #104。
在 OptiTROP-Breast01 研究中,患者按 1:1 随机接受芦康沙妥珠单抗 (sac-TMT) 治疗 (n=130) 或化疗 (n=133)。中位年龄为 51 岁;87% 存在内脏转移;26% 既往接受过 PD-1/PD-L1 抑制剂治疗;48% 在晚期阶段接受过三线或以上的化疗。
根据期中分析(数据截止日期:2023 年 6 月 21 日),已达到 PFS 主要终点,相比化疗,疾病进展或死亡的风险降低 69%(HR:0.31;95% CI:0.22 至 0.45;P<0.00001)。
根据 BICR 评估,芦康沙妥珠单抗 (sac-TMT) 的中位 PFS 为 5.7 个月(95% CI:4.3 至7.2),化疗的中位 PFS 为2.3 个月(95% CI:1.6 至2.7),6 个月的PFS 率分别为 43.4%/11.1%。在 TROP2 H 评分>200 的患者亚组中,芦康沙妥珠单抗 (sac-TMT) 的中位 PFS 为 5.8 个月,化疗的中位 PFS 为 1.9 个月(HR:0.28;95% CI:0.17 至 0.48)。
在 OS 的首次计划期中分析中(数据截止日期:2023 年 11 月 30 日,中位随访时间 10.4 个月),相比化疗,芦康沙妥珠单抗 (sac-TMT) 的 OS 显示出具有统计学意义的显著优势(HR:0.53;95% CI:0.36 至 0.78;P=0.0005);芦康沙妥珠单抗 (sac-TMT) 的中位 OS 尚未达到(95% CI:11.2 至 NE),而化疗的中位 OS 为 9.4 个月(95% CI:8.5 至 11.7)。基于 BICR 评估,芦康沙妥珠单抗 (sac-TMT)的客观缓解率 (ORR) 为43.8%,化疗的 ORR 为 12.8%。
最常见的≥3 级治疗相关不良事件 (TRAE)(芦康沙妥珠单抗(sac-TMT) /化疗)为中性粒细胞计数降低 (32.3%/47.0%)、贫血 (27.7%/6.1%) 及白细胞计数 (WBC) 降低 (25.4%/36.4%)。
3、信达生物:匹康奇拜单抗临床成功,拟报上市
5 月 28 日,信达生物宣布其自主研发的 IL-23p19 抗体注射液匹康奇拜单抗(研发代号:IBI112)在中国中重度斑块状银屑病受试者中开展的Ⅲ期临床研究(CLEAR-1)中达成主要终点和所有关键次要终点。
CLEAR-1 是目前本靶点全球唯一一个首要研究终点(16 周 PASI90)突破 80% 的银屑病三期注册临床研究。信达生物计划向 CDE 递交匹康奇拜单抗治疗银屑病的新药上市申请。
CLEAR-1(NCT05645627)是一项在中重度斑块型银屑病受试者中评估匹康奇拜单抗有效性和安全性的多中心、随机、双盲、安慰剂对照的Ⅲ期临床研究。研究共纳入 500 例受试者,按 1:2:2 的比例随机接受安慰剂、匹康奇拜单抗 200 mg(0,4,8 周 3 次给药)后,在 20 周开始接受 200 mg 或者 100 mg 每 12 周一次给药的长期维持。研究设置双主要终点,分别为第 16 周时达到银屑病面积和严重程度指数(PASI)改善≥90%(PASI 90)的受试者比例,和第 16 周时达到静态医师总体评分(sPGA)清洁(0 分)或接近清洁(1 分)的受试者比例。
研究结果显示,CLEAR-1 两个主要终点均顺利达成:第 16 周时,匹康奇拜单抗合计组 PASI 90 和 sPGA 0/1 分的受试者比例分别达到 80.3% 和 93.5%,均显著优于安慰剂组(PASI 90: 2.0%,sPGA 0/1: 13.1%; 均 P<0.0001)。这是本靶点全球唯一一个首要研究终点(16 周 PASI90)突破 80% 的银屑病三期注册临床研究,显示了匹康奇拜单抗在短期内起效明显、疗效强劲的同类最佳潜质。
至第 52 周治疗期间,匹康奇拜单抗两个剂量组达到 PASI 90 和 sPGA 0/1 分的受试者比例维持稳定。第 52 周时,200 mg 组 PASI 90 和 sPGA 0/1 分别维持在 84.9 % 及 85.9 %。这为中重度斑块状银屑病患者接受匹康奇拜单抗长期治疗的疗效获益提供了强劲的临床数据支持。
此外,匹康奇拜单抗两个剂量组所有关键次要终点均顺利达成,包括第 16 周时达到 PASI 改善≥75%(PASI 75)的受试者比例、第 16 周时达到 PASI 改善 100%(PASI 100)的受试者比例、第 16 周时达到 sPGA 清洁(0 分)的受试者比例和第 16 周时皮肤病生活质量指数(DLQI)评分为 0 或 1 分的受试者比例;匹康奇拜单抗对上述指标的改善均显著优于安慰剂组,展现出了高竞争力并基本稳健维持至第 52 周。
研究期间匹康奇拜单抗整体安全性良好,较以往临床研究未发现新的安全信号。目前研究的随访仍在持续进行中,研究相关完整数据将在未来学术会议或同行审评学术期刊上进行发表。
启动临床试验
1、奥希替尼+ Dato-DXd!第一三共/阿斯利康国内启动 III 期临床,一线治疗 EGFR 突变 NSCLC
5 月 27 日,据 CDE 临床试验登记与信息公示平台,第一三共/阿斯利康在该平台登记启动了一项 Dato-DXd(DS-1062a/德达博妥单抗) 新 III 期临床 TROPION-Lung14 研究,联合奥希替尼一线治疗 EGFR 突变阳性局部晚期或转移性 NSCLC 患者(登记号:CTR20241813)。
主要终点指标为经 BICR 评估的 PFS,次要终点包括 ORR、DOR 以及 OS 等。据 Insight 数据库显示,该项研究此前已在 ClinicalTrials.gov 平台上登记启动(登记号:NCT06350097),此次启动的是国内部分,计划国内入组 174 名受试者,国际入组 582 名,且已在 5 月 10 日完成首例受试者的入组工作。
此前 Dato-DXd 在后线治疗 EGFR 突变的 NSCLC 患者已取得积极结果。TROPION-Lung05 研究正是针对既往接受至少一种酪氨酸激酶抑制剂和至少一种含铂化疗方案(联合或不联合其他全身治疗)期间或之后出现疾病进展的晚期或转移性 NSCLC 患者。
2023 ESMO 大会上公布的该项研究数据显示,在携带 EGFR 突变的患者(n=78,患者人数最多的基因组突变)中,接受 Dato-DXd 治疗的患者 ORR 为 43.6%,DCR 为 82.1%,mPFS 为 5.8 个月,mDOR 为 7.0 个月。
在 2023 ESMO Asia 大会上则公布了该研究亚洲人群数据,137 例入组患者中有 66 例来自亚洲,其中 71% 伴有 EGFR 突变。结果显示,亚洲队列的整体 ORR 为 42.4%,中位 DOR 为 4.4 个月,DCR 为 80.3%,mPFS 为 5.4 个月。EGFR 突变患者中,ORR 达 48.9%,mDOR 为 4.4 个月,DCR 为 87.2%,mPFS 为 5.7 个月。
此外,第一三共/阿斯利康还在 ClinicalTrials.gov 平台上登记启动了一项 Dato-DXd 与奥希替尼联用二线治疗 EGFR 突变非鳞 NSCLC 的 III 期临床 TROPION-Lung15 研究(登记号:NCT06417814),计划入组 630 名受试者,预计今年 8 月完成首例受试者的入组工作。
2、恒瑞医药:SHR-1819 注射液启动 III 期临床
5 月 28 日,据 CDE 官网显示,恒瑞 SHR-1819 注射液 III 期临床试验首次公示(登记号:CTR20241927),治疗成人中重度特应性皮炎。
特应性皮炎(AD)是一种常见的慢性复发性炎症性皮肤病,其发病机制涉及遗传易感性、皮肤屏障功能障碍、皮肤菌群紊乱和免疫失调等多种因素。2019 年,我国 AD 相关疾病负担在 369 种疾病中排名第 24 位。
SHR-1819 是一种靶向人类 IL-4Rα 的重组人源化单克隆抗体,能够同时阻断 IL-4 和 IL-13 的信号传导,抑制下游炎性信号通路,从而改善疾病的炎症状态并控制疾病进展。这一机制使得 SHR-1819 在治疗 AD 及其他 2 型炎症相关性疾病中具有广泛的应用前景。
2021 年 9 月,SHR-1819 用于治疗 AD 的申请首获 NMPA 批准,随后陆续完成了 I、II 期临床,如今终于顺利启动 III 期临床试验,离上市越来越近。
而该药的在研适应症不止于此。据 Insight 数据库显示,此前 SHR-1819 用于哮喘和慢性鼻窦炎伴鼻息肉病的两项适应症也已经被获批临床,其中哮喘适应症也在 2021 年完成了 I 期临床试验。
3、安斯泰来 CD3/CLDN18.2 双抗国内启动临床
5 月 30 日,据 CDE 临床试验登记与信息公示平台显示,安斯泰来 CD3/CLDN18.2 双抗 ASP2138 国内启动 I/Ib 期研究,旨在评价在表达 CLDN 18.2 的转移性或局部晚期不可切除胃或胃食管连接部(GEJ)腺癌或转移性胰腺腺癌患者中的疗效与安全性 (登记号:CTR20241945)。
Insight 数据库显示,早在 2022 年 5 月,安斯泰来已在 ClinicalTrials.gov 上登记启动了这项临床试验(登记号:NCT05365581),计划全球入组 275 人,并于 2022 年 6 月完成了首例受试者的入组工作。本次启动的是国内部分。
ASP2138 是一款双特异性抗体,可与 CD3 和 CLDN18.2 结合。安斯泰来也是重押 CLDN18.2 的企业之一,除 ASP2138 之外还布局有单抗 zolbetuximab(佐妥昔单抗)和一款 CLDN6/CLDN-18.2/CD3 双特异性三价抗体。其中,zolbetuximab 是全球首款步入上市阶段的 CLDN18.2 单抗,今年 3 月已经在日本获批上市。在美国虽然于 1 月份收到 CRL 未能如期上市,本周也已解决第三方生产工厂问题,重新提交了上市申请。
获批临床
1、恒瑞医药:4 款创新药获批临床
本周,恒瑞陆续宣布 4 款新药或新适应症获批临床,包括 JAK1 抑制剂 SHR0302 碱凝胶、小分子新药 HRS-5346、1 类生物药 SHR-9539、GLP-1/GIP 双受体激动剂 HRS9531。
SHR0302 是高选择性的 JAK1 抑制剂,可通过抑制 JAK1 信号传导发挥抗炎和抑制免疫的生物学效应,目前口服片剂和外用软膏两种剂型正在进行多种适应症的临床开发。
片剂方面,SHR0302 目前已开展了包括类风湿关节炎、强直性脊柱炎、溃疡性结肠炎、特应性皮炎、斑秃等多领域的临床研究,其中绝大多数适应症的临床研究已进入 III 期临床试验,强直性脊柱炎、特应性皮炎和类风湿关节炎 3 项适应症的上市申请均已获得国家药监局受理。
软膏方面,用于轻中度特应性皮炎创新剂型的 SHR0302 外用软膏也已经进入 III 期临床。2018 年,恒瑞将 JAK1 抑制剂 SHR0302 许可给美国 Arcutis 公司,让创新药品惠及全球患者。
而本次获批临床的 SHR0302 碱凝胶是一种通过外用途径给药的选择性 JAK1 抑制剂,其通过外用途径给药,在制剂配方亲肤性更强,有望成为白癜风治疗的新选择。
HRS-5346 本次获批开展临床试验的适应症是脂蛋白紊乱。该药是恒瑞医药自主研发的小分子药物,临床前数据显示,HRS-5346 给药后可有效改善脂蛋白紊乱,安全性良好。恒瑞表示,脂蛋白紊乱是动脉粥样硬化性心血管疾病(ASCVD)的独立危险因素,且资料显示其患病率较高,但既往无特异性药物治疗,目前仍处于临床研究阶段,因此 HRS-5346 有望为患者提供潜在新寮费,改善其风险。
SHR-9539 是 1 类治疗用生物制品,通过诱导激活 T 细胞,使其发挥靶向杀伤多发性骨髓瘤细胞的作用,目前国内尚未有同类药物获批上市。本次该药获批适应症为多发性骨髓瘤。
HRS9531是恒瑞自主研发的 1 类创新口服 GLP-1/GIP 双受体激动剂,本次获批临床适应症为 2 型糖尿病(T2DM)和体重管理。该药也是恒瑞出海的标杆之一,包括其在内的 GLP-1 创新药组合除大中华区以外的全球范围内开发、生产和商业化的独家权利被有偿许可给设立在美国,由头部投资人贝恩资本生命科学基金、Atlas Ventures、RTW 资本等组建的新公司,恒瑞将持有该公司 19.9% 的股权,此次 GLP-1 产品组合对外许可首付款加里程碑付款累计可高达 60 亿美元。
境外创新药进展
境外部分,本周共有 22 款创新药(含改良新)研发进度推进到了新阶段,其中 1 款获批上市,8 款获批临床。
获批上市
1、Moderna:RSV mRNA 疫苗获批上市
5 月 31 日,Moderna 宣布其 RSV mRNA 疫苗获 FDA 批准上市,用于 60 岁以上老年人,商品名为 mRESVIA。该药获批之后,成为自新冠疫苗之后首款上市的 mRNA 疫苗。
RSV 是 1956 年从黑猩猩呼吸道分离出来的一种非节段性单股负链 RNA 病毒,属于副黏病毒科的肺病毒属,具有明显季节性(冬春季节为感染的高发期),常在易感人群婴幼儿和老年人中引发严重的肺炎、哮喘和支气管炎,并且可在一生中反复感染。
根据 WHO 估计,全球每年有约 3300 万 - 6000 万的儿童感染 RSV,以及大约 3% - 7% 的 60 岁以上老年人会受到 RSV 感染。
市场上针对 RSV 感染的治疗,主要包括抗体药物和 RSV 疫苗两种。其中,抗体药物主要为帕利珠单抗(Synagis)和 Beyfortus。
不过,帕利珠单抗并不完美:仅限于高危婴幼儿 RSV 预防,患者覆盖面窄,由于中和能力不高,预防效果有限、持续时间较短(保护期仅一个月),而且价格高昂(单剂成本达 1000-3000 美元)。
可即便如此,过去 10 年帕利珠单抗的销售峰值仍达到 16 亿美元。这也从侧面反映出,RSV 疫苗仍存在巨大的未被满足的临床需求,市场仍有大机遇。
因此,这一领域也成为 Moderna、GSK、辉瑞三家巨头争抢的重要领域。在 2023 年 5 月,GSK 开发的 RSVPreF3 疫苗(Arexvy/GSK 3844766A)和辉瑞的 Abrysvo(RSVpreF/PF-06928316)已经相继获 FDA 批准上市。
2、百时美施贵宝:CAR-T 疗法又一新适应症获批,用于套细胞淋巴瘤
5 月 31 日,BMS 宣布,CD19 靶向 CAR-T 疗法 Breyanzi 获 FDA 批准新适应症,用于既往接受过至少 2 线系统性疗法的复发性或难治性套细胞淋巴瘤(MCL)成人患者,包括曾接受过 BTK 抑制剂治疗的患者。
这是该药第 5 次取得 FDA 批准。本月中(5 月 16 日),该药才刚刚获批用于既往接受过二线及以上系统疗法的复发/难治性滤泡性淋巴瘤。
本次批准是基于 TRANSCEND NHL 001 试验的 MCL 队列结果。在接受 Breyanzi 治疗并进行疗效评估的患者(n=68)中,ORR 为 85.3%(95% CI:74.6-92.7),其中 67.6% 的患者达到 CR(95% CI:55.2-78.5)。超过一半(51.4%;95% CI:37.5-63.7)的缓解者在 12 个月时仍保持缓解,38.8%(95% CI:25-52.4)的缓解者在 18 个月时仍保持缓解。
3、礼来:RET 抑制剂新适应症再获加速批准,可用于 12 岁以下儿童
5 月 29 日,FDA 官网显示,礼来的 RET 激酶抑制剂塞普替尼(Selpercatinib,商品名 Retevmo)获得加速批准,用于治疗 2 岁及以上的儿童患者,涵盖 RET 突变晚期或转移性甲状腺髓样癌(MTC)、RET 基因融合晚期或转移性甲状腺癌、RET 基因融合,接受全身性治疗后疾病进展或没有满意替代治疗方案的局部晚期或转移性实体瘤。
该药此前已经获批用于成人和 12 岁及以上儿童的甲状腺癌适应症,以及成人的实体瘤适应症。本次获批,其适应症的年龄阶段得以再一次放宽,也使 12 岁以下儿童得以获得这款新的靶向治疗方案。
塞普替尼是一种同类首创,高选择性和抑制活性的小分子 RET 抑制剂,具有中枢神经系统(CNS)活性,可抑制多种 RET 变异。该药也是全球首款获批的高选择性 RET 抑制剂,通过抑制异常 RET 激酶的活性而发挥作用。
塞普替尼已先后获得美国 FDA 三项突破性疗法认证和优先审评审批资格,并于 2020 年 5 月经美国 FDA 批准上市(美国商品名 Retevmo®),用于治疗转染重排基因(RET)融合阳性转移性非小细胞肺癌(NSCLC)的成年患者,和需要系统性治疗的携带 RET 突变的晚期或转移性甲状腺髓样癌成人和 12 岁及以上的儿童患者,以及需要系统性治疗和放射性碘治疗(如适用)难治的 RET 融合阳性的晚期或转移性甲状腺癌成人和 12 岁及以上的儿童患者。
2022 年 9 月,FDA 批准塞普替尼作为首个且唯一不限癌种 RET 抑制剂,用于 RET 基因融合的晚期或转移性实体瘤成人患者,同时常规批准该药用于 RET 融合阳性的局部晚期或转移性非小细胞肺癌(NSCLC)成人患者。
2022 年 10 月,塞普替尼也在国内获批上市。礼来已与信达达成合作,共同进行国内市场的商业化。
申报上市
1、安斯泰来:再次提交 CLDN18.2 单抗上市申请
5 月 31 日,安斯泰来宣布向 FDA 重新递交 CLDN18.2 单抗 Zolbetuximab 的上市申请,PDUFA 日期为 2024 年 11 月 9 日。
Zolbetuximab 是全球首款申报上市的同类药物,今年 3 月已经在日本获批。1 月份,由于第三方生产工厂问题,使之不能在原定的 PDUFA 决定日期得到审评结论,憾收 FDA 的 CRL,如今终于重新提交上市申请。
其 NDA 是基于两项 III 期临床 SPOTLIGHT 和 GLOW 的研究结果。
SPOTLIGHT 研究探索了 Zolbetuximab 联合 mFOLFOX6(奥沙利铂、亚叶酸 + 氟尿嘧啶联合方案)一线治疗 CLDN18.2 阳性、HER2 阴性局部晚期不可切除或转移性胃或胃食管交界处腺癌的疗效和安全性。
另一项 III 期临床 GLOW 研究则评估了 Zolbetuximab 联合 CAPOX(卡培他滨 + 奥沙利铂)一线治疗胃癌的疗效。
2、罗氏:PI3Kα 抑制剂申报上市,获 FDA 优先审评资格
5 月 29 日,罗氏宣布 Inavolisib 在美国的上市申请获得优先审评资格,与哌柏西利和氟维司群联用治疗 PIK3CA 突变、HR+、HER2- 的局部晚期或转移性乳腺癌成人患者,这些患者在完成辅助内分泌治疗后 12 个月内复发。
PDUFA 日期为 2024 年 11 月 27 日。
Inavolisib 是一种创新口服靶向治疗药物,能为 HR 阳性、PIK3CA 突变的乳腺癌患者提供良好的耐受性、持久的疾病控制并有可能改善预后。PIK3CA 突变率约 40%,在乳腺癌患者较常见但常常被忽视。
Inavolisib 设计用于降低治疗的总体毒性,有别于其他的 PI3K 抑制剂,它在体外对 PI3Kα 的抑制有高效力和特异性,同时有独特的作用机制可降解 PI3Kα 突变体。
去年 12 月,罗氏宣布 III 期临床试验 INAVO120 研究取得阳性结果。表明inavolisib 联合哌柏西利和氟维司群,可作为 PIK3CA 突变的 HR 阳性、HER2 阴性、内分泌耐药的局晚或转移性乳腺癌患者的一线治疗选择。
此次获得 FDA 优先审查资格主要基于 III 期临床 INAVO120 研究的积极结果。
INAVO120 研究(NCT04191499)旨在在 PIK3CA 突变、HR 阳性、HER2 阴性局部晚期或转移性乳腺癌患者中,评价 Inavolisib 联合哌柏西利和氟维司群对比安慰剂联合哌柏西利和氟维司群治疗有效性和安全性。患者在辅助内分泌治疗期间或治疗完成后 12 个月内发生疾病进展,且既往针对转移性疾病未接受过系统治疗。
该研究入组 325 例患者,随机分配到研究组或对照组治疗。主要终点是研究者评估的无进展生存期,定义是随机至疾病进展或由于任何原因死亡的时间。次要终点包括总生存期、客观缓解率和临床获益率。
结果显示,与哌柏西利和氟维司群的对照组相比,Inavolisib 联合哌柏西利和氟维司群治疗组将疾病进展或死亡风险降低了 57%,中位 PFS 为 15.0 个月 VS 7.3 个月。虽然 OS 数据尚不成熟,但已观察到明显的积极趋势,后续将继续进行下一次分析。安全性方面,Inavolisib 联合治疗耐受性良好,不良反应与已知安全性特征一致,未观察到新的安全信号。
Inavolisib 目前在 PIK3CA 突变局部晚期或转移性乳腺癌中开展了 3 项 III 期临床研究,除 INAVO120 研究外,还在开展:1)联合氟维司群 vs.alpelisib 联合氟维司群治疗 CDK4/6 抑制剂联合内分泌经治的 HR 阳性/HER2 阴性乳腺癌患者的 INAVO121 研究;2)联合帕妥珠曲妥珠单抗注射液(皮下注射)(Phesgo®)vs.Phesgo 作为一线 HER2 阳性乳腺癌维持治疗的 INAVO122 研究。
在国内,据 CDE 显示,Inavolisib 也已被纳入优先审评。
3、Zymeworks:HER2 双抗在美国首次申报上市,治疗胆道癌
5 月 30 日,Jazz Pharmaceuticals 和 Zymeworks 共同宣布,美国 FDA 已接受其在研 HER2 双抗 zanidatamab 的 BLA 申请并授予优先审评资格,用以治疗经治、不可切除、局部晚期或转移性 HER2 阳性胆道癌(BTC)患者,PDUFA 日期在 2024 年 11 月 29 日。
如若获批,该药将成为 FDA 批准用于治疗 HER2 阳性局部晚期或转移性 BTC 患者的首个 HER2 靶向药物。同时,也会是首款获批上市的 HER2 双抗。
本次申请主要基于 HERIZON-BTC-01 临床试验数据。该试验是一项 2b 期研究,评估 zanidatamab 在经治 HER2 阳性 BTC 患者中的疗效与安全性。试验主要终点是通过独立中央审评(ICR)检视在队列 1 中的确认客观缓解率(cORR)。
在 6 月 2 日(今日)刚刚举行的 ASCO 年会上,两家公司刚刚公布了 HERIZON-BTC-01 的长期随访结果。分析显示,既往曾接受吉西他滨治疗的 HER2 阳性、局部晚期不可切除或转移性胆道癌(BTC)患者,在接受 zanidatamab 治疗后的中位 OS 达 15.5 个月,远超过过往类似患者群体 6 - 9 个月的生存期。
截至 2023 年 7 月 28 日,在队列 1(n=80)的 BTC 患者中,患者的确认客观缓解率(cORR)为 41.3%(n=33,95% CI:30.4-52.8)。其中,不同 HER2 表达患者的亚组分析显示,IHC 3+患者的 cORR 达 51.6%(95% CI:38.6-64.5),而 IHC 2+患者的 cORR 为 5.6%(95% CI:0.1-27.3)。
患者的中位 OS 为 15.5 个月(95% CI:10.4,18.5),其中 IHC 3+患者的中位 OS 为 18.1 个月(95% CI:12.2,23.2),IHC 2+患者的中位 OS 为 5.2 个月(95% CI:3.1,10.2)。
另外,值得一提的是,百济神州曾在 2018 年度就与 Zymeworks 达成合作,在亚太地区开发 HER2 双抗 ZW25(即本次申报上市的药物)和 HER2 双抗 ADC ZW49。
临床试验结果
1、第一三共/阿斯利康:TROPION-Lung01 研究结果披露,Dato-DXd 展现具有临床意义的 OS 改善
5 月 27 日,第一三共与阿斯利康共同宣布,III 期临床 TROPION-Lung01 研究更新结果表明,相较于多西他赛,Dato-DXd 在既往至少接受过一线治疗的局部晚期或转移性非鳞 NSCLC 患者中,在 OS 方面表现出有临床意义的改善。
Dato-DXd 是由第一三共和阿斯利康共同开发的 TROP2 ADC。
TROPION-Lung01 研究是 Dato-DXd 在肺癌领域开展的首个 III 期临床研究,旨在评估 Dato-DXd 单药对比多西他赛在既往至少接受过一次治疗的在携带或不携带驱动基因改变的局部晚期或转移性 NSCLC 患者的有效性和安全性。主要终点为 BICR 评估的 OS 和 PFS。
去年已经达到 PFS 主要终点。在 2023 ESMO 大会上公布的 PFS 结果显示,Dato-DXd 在总体试验人群中展示出具有统计学意义的的 PFS 改善,尤其在非鳞状 NSCLC 患者中 PFS 时间延长超过 50%(5.5 个月 v.s. 3.6 个月)。且在 TROPION-Lung01 研究中,入组人群与真实世界发病率保持一致,约 75% 的患者为非鳞状 NSCLC。
而此次更新结果表明,与化疗相比,Dato-DXd 也可为晚期非鳞状 NSCLC 患者带来具有临床意义的 OS 改善。安全性方面,与既往分析结果一致,未发现新的安全性问题,也未观察到其他 5 级间质性肺部疾病事件。
针对既往接受过全身治疗的局部晚期或转移性非鳞状 NSCLC 成人患者,Dato-DXd 在美国和欧盟地区正处于上市申请中。
2、吉利德:TROP2 ADC III 期确证性临床试验失败,治疗尿路上皮癌
5 月 30 日,吉利德宣布,Trop2 ADC 药物戈沙妥珠单抗(Trodelvy)治疗晚期尿路上皮癌(mUC)三期临床 TROPiCS-04 未能达到 OS 主要终点。
戈沙妥珠单抗是全球首款获批上市的 TROP2 ADC,最初获批在 2020 年度。此前该药已经在三阴乳腺癌、尿路上皮癌、HR+/HER2-乳腺癌上取得监管批准。2023 年度,该药全球销售额首次突破 10 亿美元。以此为代表的 TROP2 靶点也是 ADC 热捧的 TOP3 靶点,第一三共/阿斯利康的 Dato-DXd 和科伦博泰/默沙东的 SKB264/MK-2870 都紧随其后。
吉利德表示,将对本次 III 期临床试验错失 OS 主要终点的相关数据进行分析,并与 FDA 进行讨论。
这并不是 Trodelvy 失败的第一项研究,在今年 1 月 22 日,Trodelvy 对比多西他赛治疗经治转移性或晚期 NSCLC 患者的 III 期临床 EVOKE-01 研究也未能达到 OS 主要终点。
四川省医药保化品质量管理协会举办2026
当前,制药行业正处于合规升级与绿..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..关于收取2026年度会费的通知
各会员单位: 在过去的一年里,..四川省医药保化品质量管理协会党支部召
按照省市场监督管理局社会组织联合..


