

预测性生物标志物是实现精准医疗的重要条件,随着药物-伴随诊断(CDx)联合开发模式的引入,在全球最大医药市场,美国监管机构FDA批准的伴随诊断产品数量稳步增加。获得批准的伴随诊断检测产品主要涉及到不同的血液和肿瘤药物。如果没有准确可靠的伴随诊断检测,这些药物将失去其价值。
FDA批准数量剧增
2014年,FDA发布了关于《体外伴随诊断试剂指导原则》的监管指导文件,将这种类型的检测定义为体外诊断设备(IVD),可提供对安全有效使用相应治疗产品至关重要的信息。伴随诊断的使用在检测使用说明和相应治疗产品的标签中都有规定,包括治疗产品的任何通用等价物的标签。这意味着在美国使用伴随诊断检测进行测试是强制性的,并且必须在向患者开具特定治疗产品处方之前进行。
FDA自1998年首次批准伴随诊断产品以来,截止2020年底,获批总数已达到44种。
2010年(包括2010年)之前,FDA仅批准了9种伴随诊断检测产品,此后的10年,FDA批准了35种。几乎所有这些伴随诊断检测产品都与血液学和肿瘤学药物相关,与同一时期FDA批准的靶向抗癌疗法数量增加遥相呼应。
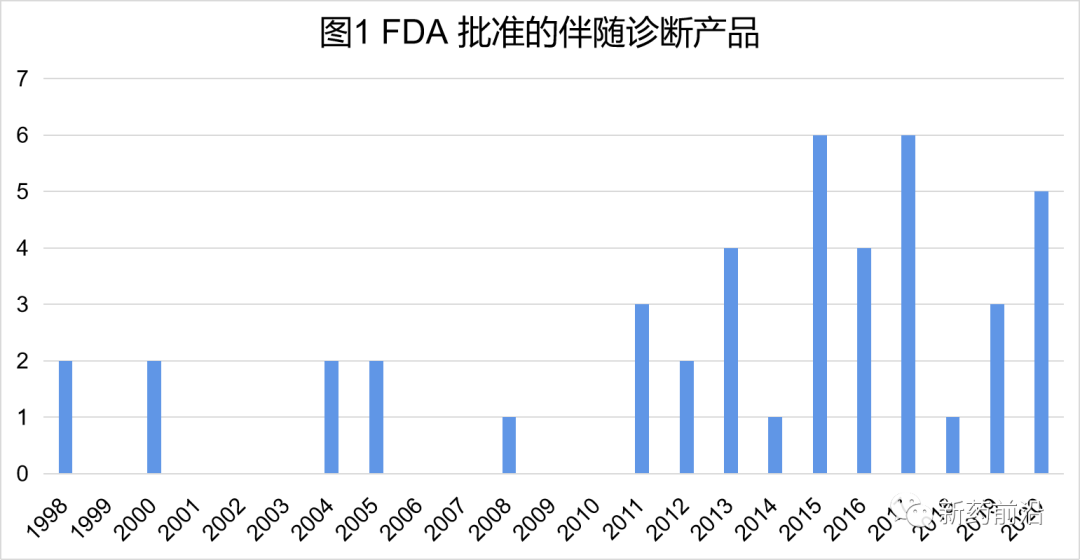
过去20多年来观察到的趋势显示,在抗癌药物开发中更频繁地使用药物-伴随诊断检测产品联合开发模式越来越普遍。
从FDA批准的伴随诊断检测相关的生物标志物、药物和适应症来看。迄今为止,药物-伴随诊断检测产品联合开发模式主要基于“一种药物一种生物标志物”的。尽管与伴随诊断产品同时获批的小分子靶向药物占据绝对优势,而需要生物标志物检测的单抗药物也在不断增加。
诺华的地拉罗司(Exjade)是唯一的非抗癌药物,该药是一种铁螯合剂,适用于非输血依赖型地中海贫血症(NTDT)患者慢性铁过载的治疗。为筛选Exjade适用人群,FDA授予FerriScan作为肝铁浓度(LIC)测量影像诊断方法的上市销售许可。FDA之前批准FerriScan用于肝铁浓度测量,不过,Exjade临床研究中用它进行治疗患者筛选和治疗管理,因此确立了其作为Exjade安全和有效使用所必需的伴随成像诊断方法。
技术平台快速革新
从不同伴随诊断检测的分析平台来看,免疫组织化学(IHC)和原位杂交(ISH)是主导技术,直到2011年,第一个基于聚合酶链反应(PCR)方法的伴随诊断获得批准,即cobas 4800 BRAF V600突变测试产品(罗氏)。该产品是一种用于黑色素瘤患者BRAF V600E突变的检测,这些患者可能是维罗非尼(Zelboraf)治疗的候选者。迄今为止,PCR技术构成了最大的伴随诊断检测类别;共有16种基于PCR的检测产品已获得FDA批准,占FDA批准的所有伴随诊断检测产品的36%。
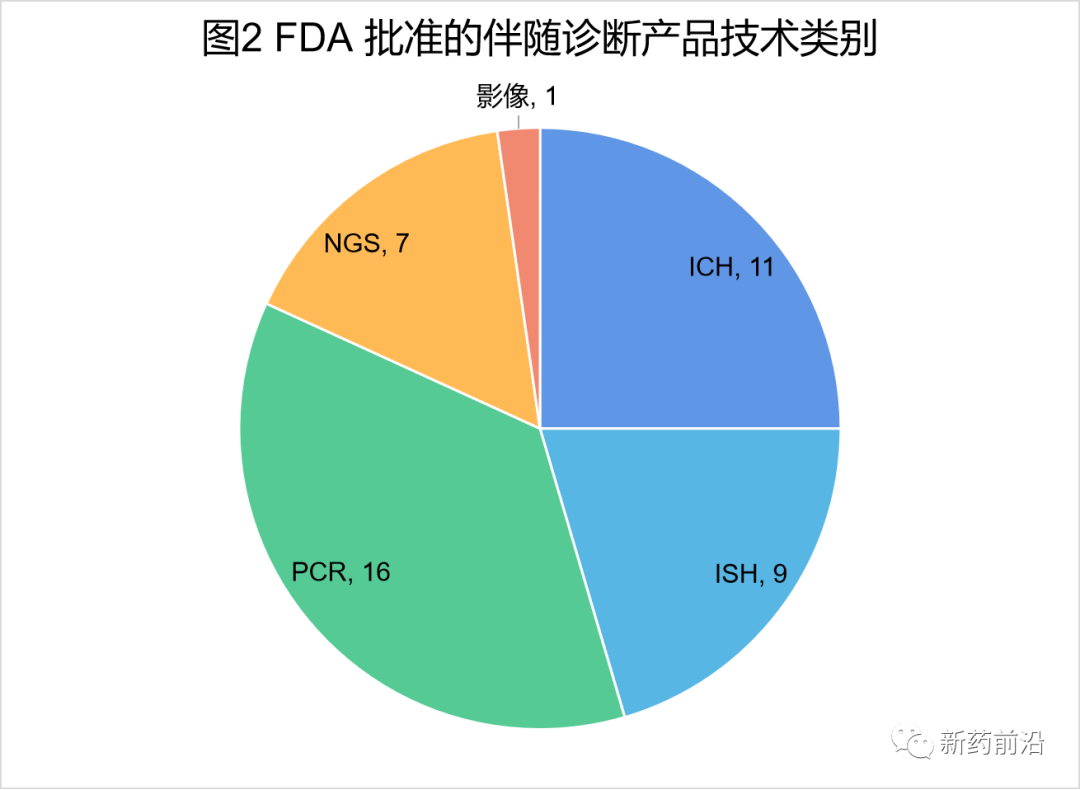
在过去五年中,新一代测序(NGS)也已成为新的伴随诊断平台。FDA批准的第一个基于该技术的检测方法是FoundationFocus伴随诊断BRCA检测(Foundation Medicine公司),这是一种检测卵巢癌患者肿瘤组织中BRCA1和BRCA2改变的检测方法,这些患者可能是接受鲁卡帕里布(Rubraca)治疗的候选者。目前,七种基于NGS不同的伴随诊断检测产品已获得FDA批准。
在过去的几年里,我们看到FDA于2018年批准了广谱抗癌药物,拜耳/Loxo Oncology公司的larotrectinib(Vitrakvi)用于治疗携带NTRK基因融合的成年和儿童局部晚期或转移性实体瘤患者,不需考虑癌症的发生区域。直到2020年,FDA才批准FoundationOne伴随诊断产品用作首个伴随诊断方法,以帮助鉴定可能适合使用Vitrakvi治疗的神经营养受体酪氨酸激酶(NTRK)基因融合阳性患者。FoundationOne是FDA批准的针对所有实体瘤的综合基因组分析(CGP)测试产品。
值得一提的是,在美国,大多数伴随诊断检测是高风险设备,归类为III类,需要提交上市前申请(PMA)。

后记
在我国,国家药监局今年4月7日发布《基于同类治疗药物的肿瘤伴随诊断试剂说明书更新与技术审查指导原则》。市场监管总局已经在今年7月22日出台《体外诊断试剂注册与备案管理办法》,并2021年10月1日起施行。伴随诊断市场发展的政策支持日益完善。
高端分子诊断设备平台目前还严重依赖进口,随着国内该领域长期的技术积累和创新,期望尽早实现技术突破和国产替代。
党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..党支部开展《树立和践行正确政绩观 促
2026年6月26日,由四川省医药保化..2026年第一期药品生产企业拟新任质量受
应我省部分药品生产企业需要新增设..四川省医药保化品质量管理协会召开第七
2026年6月8日,四川省医药保化品质..关于召开第七届九次理事会暨会长办公会
各相关单位: 经研究,四川省医..关于举办四川省药品生产企业拟新任质量
各相关企业: 新修订的《中华人..四川省医药保化品质量管理协会党支部
2026年4月8日上午,省医药保化品质..120亿美元押中!诺华AOC临床告捷
6月11日,诺华宣布其针对面肩肱型..


